
Аутофагосома — Autophagosome — qaz.wiki
Аутофагосома представляет собой сферическую структуру с двойным слоем мембран. Это ключевая структура в макроаутофагии , системе внутриклеточной деградации цитоплазматического содержимого (например, аномальных внутриклеточных белков, избыточных или поврежденных органелл, вторгающихся микроорганизмов). После образования аутофагосомы доставляют цитоплазматические компоненты к лизосомам . Наружная мембрана аутофагосомы сливается с лизосомой с образованием автолизосомы . Гидролазы лизосомы разрушают доставляемое аутофагосомами содержимое и его внутреннюю мембрану.
Образование аутофагосом регулируется генами, которые хорошо законсервированы от дрожжей до высших эукариот. Номенклатура этих генов различалась от бумаги к бумаге, но в последние годы она была упрощена. Семейства генов, ранее известные как APG, AUT, CVT, GSA, PAZ и PDD, теперь объединены в семейство ATG (родственное AuTophaGy).
Размер аутофагосом у млекопитающих и дрожжей различается . Аутофагосомы дрожжей имеют размер около 500-900 нм, а аутофагосомы млекопитающих больше (500-1500 нм). В некоторых примерах клеток, таких как эмбриональные стволовые клетки , эмбриональные фибробласты и гепатоциты , аутофагосомы видны с помощью светового микроскопа и могут рассматриваться как кольцеобразные структуры.
Формирование аутофагосом
Начальный этап аутофагосомного образования омегасомы на эндоплазматическом ретикулуме , за которым следует удлинение структур, называемых фагофорами.
Формирование аутофагосом контролируется генами Atg через комплексы Atg12-Atg5 и LC3. Конъюгат Atg12-Atg5 также взаимодействует с Atg16 с образованием более крупных комплексов. Модификация Atg5 с помощью Atg12 необходима для удлинения исходной мембраны.
После образования сферической структуры комплекс ATG12 — ATG5 : ATG16L1 диссоциирует от аутофагосомы. LC3 расщепляется протеазой ATG4 с образованием цитозольного LC3. Расщепление LC3 необходимо для терминального слияния аутофагосомы с ее мембраной-мишенью. LC3 обычно используется в качестве маркера аутофагосом в иммуноцитохимии , поскольку он является важной частью везикулы и остается связанным до последнего момента перед слиянием. Сначала аутофагосомы сливаются с эндосомами или везикулами, происходящими из эндосом. Эти структуры затем называют амфисомами или промежуточными аутофагическими вакуолями. Тем не менее, эти структуры содержат эндоцитические маркер даже небольшие лизосомальные белки , такие как катепсин D .
Расщепление LC3 необходимо для терминального слияния аутофагосомы с ее мембраной-мишенью. LC3 обычно используется в качестве маркера аутофагосом в иммуноцитохимии , поскольку он является важной частью везикулы и остается связанным до последнего момента перед слиянием. Сначала аутофагосомы сливаются с эндосомами или везикулами, происходящими из эндосом. Эти структуры затем называют амфисомами или промежуточными аутофагическими вакуолями. Тем не менее, эти структуры содержат эндоцитические маркер даже небольшие лизосомальные белки , такие как катепсин D .
Этот процесс аналогичен у дрожжей, однако названия генов различаются. Например, LC3 у млекопитающих — это Atg8 у дрожжей, а аутофагосомы образуются из преаутофагосомной структуры (PAS), которая отличается от структур-предшественников в клетках млекопитающих. Преаутофагосомная структура у дрожжей описывается как комплекс, локализованный около вакуоли. Однако значение этой локализации неизвестно. Зрелые аутофагосомы дрожжей сливаются непосредственно с вакуолями или лизосомами и не образуют амфисомы, как у млекопитающих.
В созревании аутофагосом дрожжей также участвуют другие известные игроки как Atg1 , Atg13 и Atg17. Atg1 представляет собой киназу, активируемую при индукции аутофагии. Atg13 регулирует Atg1, и вместе они образуют комплекс под названием Atg13: Atg1, который получает сигналы от мастера восприятия питательных веществ — Tor. Atg1 также важен на поздних стадиях формирования аутофагосом.
Функция в нейронах
В нейронах аутофагосомы образуются на кончике нейрита и созревают (подкисляются) по мере продвижения к телу клетки по аксону . Этот аксональный транспорт нарушается, если гентингтин или его взаимодействующий партнер HAP1 , которые колокализуются с аутофагосомами в нейронах, истощаются.
Рекомендации
<img src=»//en.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1×1″ alt=»» title=»»>Аутофагия
Термин «аутофагия», в переводе с греческого означающий «самопоглощение», отражает один из важнейших процессов саморегуляции, в ходе которого из функционирующей клетки удаляются как собственные дефектные органеллы и цитозольные белки, так и инородные, патогенные структуры.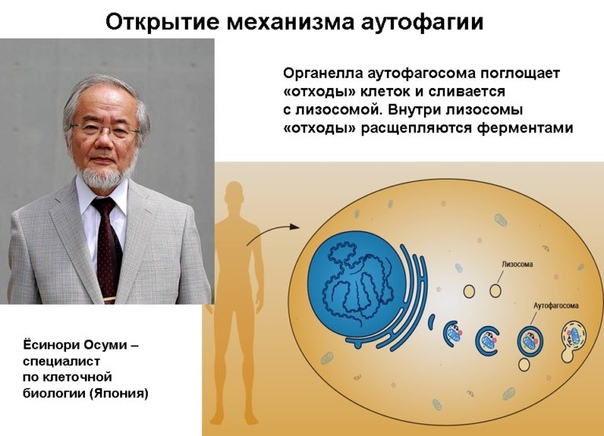
В 1960-х годах исследователи впервые обнаружили, что клетка, находящаяся в состоянии энергетической недостаточности или окислительного стресса может разрушать свое собственное содержимое, заключая его в мембраны (фагофоры), постепенно сливающиеся и образующие везикулы (аутофагосомы), которые затем транспортируются в лизосомы для деградации. Этот процесс позже будет назван макроаутофагией.
3 октября 2016 года Нобелевская премия по физиологии и медицине была присуждена Yoshinori Ohsumi за «открытие механизмов аутофагии». Исследователь культивировал мутировавшие дрожжи, лишенные ферментов лизосомальной дегенерации, одновременно лишая их питательного субстрата для стимуляции процессов аутофагии. В лизосомах таких дрожжей были обнаружены крупные скопления аутофагосом, позже Ohsumi выделил ключевые гены, вовлеченные в этот процесс.
Сегодня выделяют три типа аутофагии: макроаутофагия, микроаутофагия и аутофагия, опосредованная белками-шаперонами. Однако именно механизмы макроаутофагии наиболее изучены, а их нарушение ассоциировано с развитием целого ряда заболеваний.
Во многих исследованиях уже продемонстрирована роль механизмов аутофагии в подавлении опухолевой прогрессии, функционировании врожденного и адаптивного иммунитета, регуляции путей системного воспаления, процессах нейродегенерации и старения. Способность клеток к аутофагии может быть скомпрометирована внешними или внутренними факторами, что способствует их преждевременной гибели, и возможно, сокращает продолжительность жизни.
Сегодня способы влияния на генетические и молекулярные механизмы аутофагии рассматриваются как стратегия будущего в терапии и предотвращении развития ряда заболеваний. Наиболее мощными выявленными индукторами процессов аутофагии являются голодание и ограничение количества поступающих с пищей калорий. На этой концепции построены набирающие популярность режимы питания, например такие, как интервальное голодание (intermittent fasting), когда все приемы пищи происходят в течение 8-часового временного окна, за которым следует 16 часов без употребления каких-либо продуктов.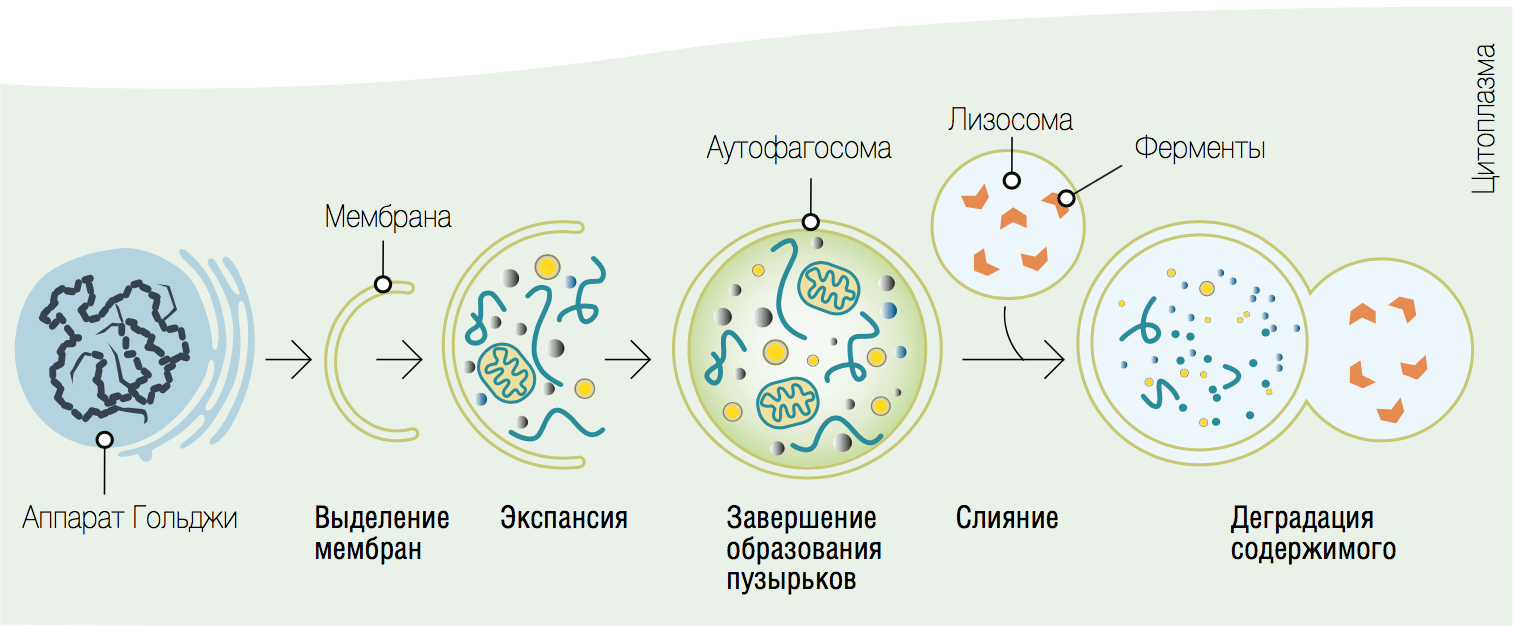
Существуют различные экспериментальные модели интервального голодания, применение которых к лабораторным животным приводило к улучшению когнитивных функций, задержке процессов старения, а также было продемонстрировано вероятное противовоспалительное действие такого типа питания. Отмечалось также увеличение разнообразия состава кишечной микробиоты после соблюдения Рамадана, который можно рассматривать как одну из моделей периодического голодания. Дальнейшие открытия в этой области могут существенно изменить наше представление о здоровом образе жизни.
Источники:
- Ying Yang1 ● Daniel J. Klionsky1. Autophagy and disease: unanswered questions. Cell Death & Differentiation. Received: 21 October 2019 / Revised: 3 December 2019 / Accepted: 6 December 2019
- Beth Levinea,b,c,1 and Daniel J. Klionskyd. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A. 2017 Jan 10
- Beth Levine1,2,* and Guido Kroemer3,4,5,*. Autophagy in the Pathogenesis of Disease. Cell. Author manuscript; available in PMC 2009 Jun 16.
- Mary-Catherine Stockman, RD, LDN,1 Dylan Thomas, MD,1 Jacquelyn Burke, MS, RD,2 and Caroline M. Apovian, MD. Intermittent Fasting: Is the Wait Worth the Weight? Curr Obes Rep. Author manuscript; available in PMC 2019
- Structural changes in gut microbiome after Ramadan fasting: a pilot study C. Ozkul , M. Yalinay , T. Karakan
____________
Читайте также:
За что дали Нобелевскую премию по медицине: комментарий эксперта
Автор фото, NOBEL FOUNDATION
Премию ему вручили за исследования механизмов аутофагии — так называется процесс «самопоедания» клеток. Осуми является молекулярным биологом и работает в Токийском технологическом институте.
Вот как прокомментировал это известие Константин Северинов, специалист в области молекулярной биологии (регуляция транскрипции генов бактерий), профессор Сколковского института науки и технологий:
Явление аутофагии известно довольно давно, и суть его в том, что клетки должны не только расти, но также должны какие-то свои части время от времени разлагать, такой контроль качества, когда составные части клетки стареют и должны быть заменены, а перед этим уничтожены. Это происходит в клетках каждого организма, но генетическая подоплека этого процесса была совершенно неизвестна, а само явление, когда в клетках наблюдали такие пузырьки — их называют вакуоли, аутофагосомы, в которых видели фрагменты вроде бы нормальных частей клетки, — это наблюдали еще в 50-х годах ХХ века.
А то, что лауреат сделал, — он в конце 80-х-начале 90-х годов перешел на дрожжи вместо того, чтобы изучать клетки человека, они являются замечательным модельным объектом для изучения — с точки зрения сложности они похожи на клетки человека, но работать с ними гораздо легче с точки зрения генетики.
Проблема в том, что явление аутофагии в клетках дрожжей не было известно, они очень маленькие, их неудобно смотреть в микроскопе. Тем не менее, с помощью каких-то красивых экспериментальных подходов Осуми удалось показать, что аутофагия в дрожжах существует, а дальше был вопрос техники — выявить те гены, мутации в которых нарушают этот процесс. А потом оказалось, что у человека гены, ответственные за аутофагию, — такие же, как у дрожжей. Таким образом были идентифицированы все те гены, которые необходимы нам, чтобы этот процесс у нас шел.
Принципиальная важность открытия в том, что открыт и на генетическом уровне охарактеризован важнейший клеточный процесс. С практической точки зрения, если клетка умеет переваривать себя, если у нее есть специальный генетически контролируемый процесс или каскад реакций, который позволяет это делать, то, например, если бы вы смогли активировать такой процесс в раковых клетках, которые бессмертны, то можно таким образом пытаться бороться с раком. Если можно процесс аутофагии индуцировать каким-то образом в раковых клетках, то наверное, можно пытаться их таким образом убить.
Если можно процесс аутофагии индуцировать каким-то образом в раковых клетках, то наверное, можно пытаться их таким образом убить.
Этот же процесс аутофагии вовлечен в борьбу организма с инфекционными заболеваниями, — когда бактерия попадает в клетку, клетка борется с ней, создавая те самые вакуоли и используя те же самые белки, которые вовлечены в аутофагию.
У пожилых людей процесс аутофагии участвует в старении, потому что он не очень хорошо работает и у нас накапливается шлак. А если бы можно было этот процесс активировать и сделать это контролируемо и правильно, я боюсь говорить, что можно получить омоложение, но как принципиальный подход, можно про это думать.
В России количество ученых, занятых биомедициной, мало. С одной стороны, странно, конечно, говорить про 40-е годы, но Советский Союз — одна из немногих стран, где генетика подверглась систематическому разгрому со стороны государства, и последствия этого, безусловно, ощущаются. Скажем так, не стимулировало то, чтобы наша страна стала передовой в этой области.
Кроме того, отток ученых начала 90-х годов привел к тому, что качество научных исследований в России упало. Ну и самое главное, что такого рода исследования требуют определенной инфраструктуры, налаживания поставок реагентов, животных для опытов, оборудования. Казалось бы, банальные вещи.
Так вот, Россия — одна из стран, где проблема бесперебойной доставки вещей, необходимых для такого рода исследований, до сих пор не решена.
О ВЗАИМОСВЯЗИ АУТОФАГИИ И АПОПТОЗА В ГИБЕЛИ КЛЕТОК КАРЦИНОМЫ ЛЕГКОГО А549, ИНДУЦИРОВАННОЙ CD437 | Вартанян
1. Wolf G. A history of vitamin A and retinoids. FASEB J 1996;10:1102–1107. DOI: 10.1096/fasebj.10.9.8801174.
2. Goodman G.E., Alberts D.S., Meyskens F.L. Retinol, vitamins, and cancer prevention: 25 Years of learning and relearning.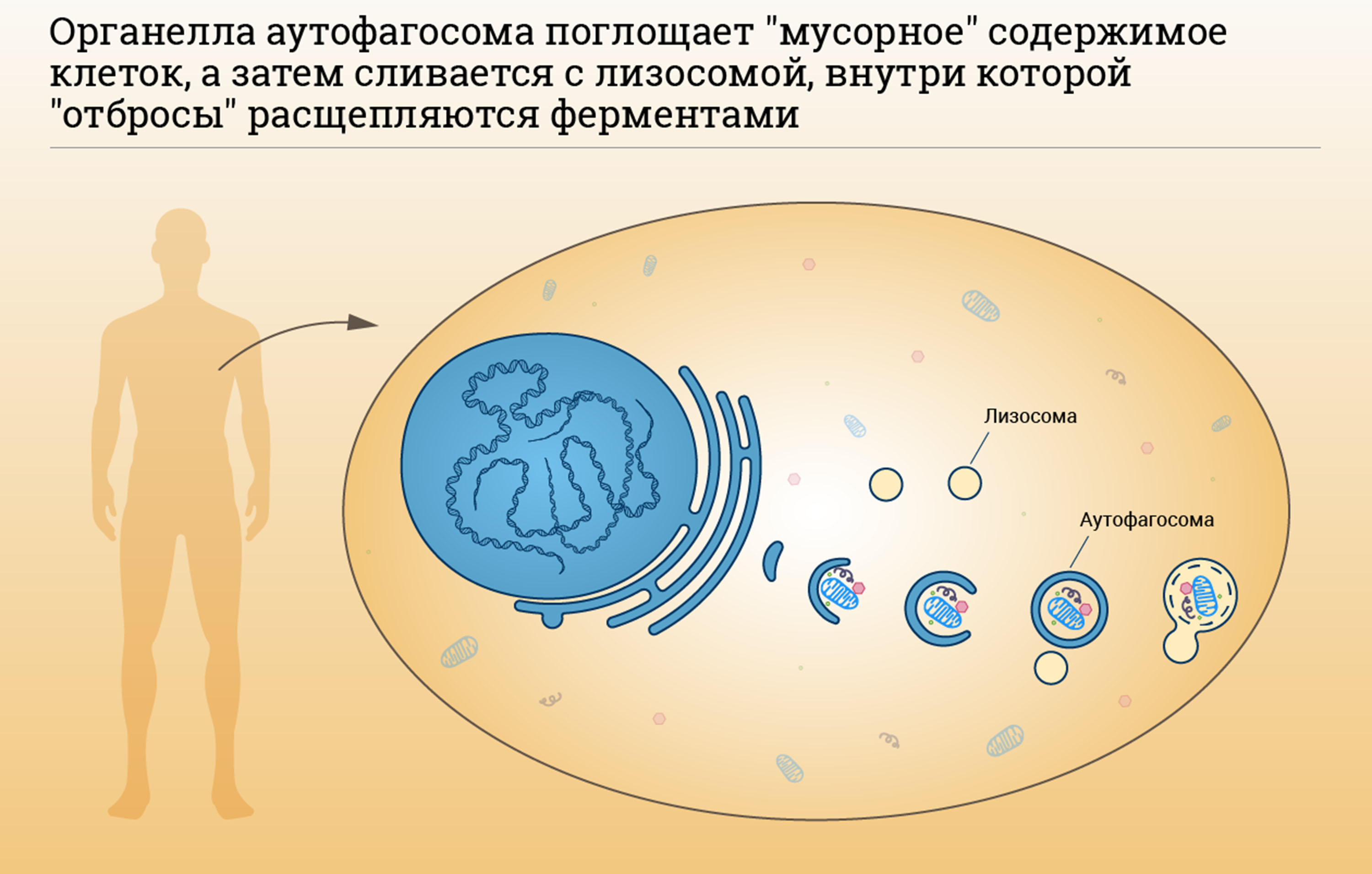
3. Smith W.E., Yazdi E., Miller L. Carcinogenesis in pulmonary epithelia in mice on different levels of vitamin A. Environ Res 1972;5:152–63. DOI: 10.1016/0013-9351(72)90030-8.
4. Zhao X., Spanjaard R.A. The apoptotic action of the retinoid CD437/AHPN: diverse effects, common basis. J Biomed Sci 2003;10(1):44–9. DOI: 10.1007/BF02255996.
5. Chen J.Y., Clifford J., Zusi C. Two distinct actions of retinoid-receptor ligands. Nature 1996;382(6594):819–22. DOI: 10.1038/382819a0.
6. Garattini E., Gianni M., Terao M. Retinoid related molecules an emerging class of apoptotic agents with promising therapeutic potential in oncology: pharmacological activity and mechanisms of action. Curr Pharm Des 2004;10(4):433–48. DOI: 10.2174/1381612043453351.
7. White E. The role for autophagy in cancer. J Clin Invest 2015;125(1):42–6. DOI: 10.1172/JCI73941.
8. Levy J.M., Towers C.G., Thorburn A. Targeting autophagy in cancer. Nature Reviews Cancer 2017;17(9):528–42. DOI: 10.1038/nrc.2017.53.
9. Bollag W., Isnardi L., Jablonska S. Links between pharmacological properties of retinoids and nuclear retinoid receptors. Int J Cancer 1997;70:470–2. PMID: 9033657.
10. Zhao X., Demary K., Wong L., Vaziri C. Retinoic acid receptor-independent mechanism of apoptosis of melanoma cells by the retinoid CD437 (AHPN). Cell Death Differ 2001;8(9):878–86. DOI: 10.1038/sj.cdd.4400894.
Cell Death Differ 2001;8(9):878–86. DOI: 10.1038/sj.cdd.4400894.
11. Shyu R.Y., Lin D.Y., Reichert U. Synthetic retinoid CD437 induces celldependent cycle arrest by differential regulation of cell cycle associated proteins. Anticancer Res 2002;22(5):2757–64. PMID: 12529993.
12. Lotan R. Receptor-independent induction of apoptosis by synthetic retinoids. J Biol Regul Homeost Agents 2003;17(1):13–28. PMID: 12757019.
13. Saxton R.A, Sabatini D.M. mTOR Signaling in Growth, Metabolism, and Disease. Сell 2017;168:960–76. PMID: 28283069.
14. Karimi Roshan M., Soltani A., Soleimani A. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019;165:229–34. DOI: 10.1016/j.biochi.2019.08.003.
15. Guo X., Wang X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res 2009;19(1):71–88. DOI: 10.1038/cr.2008.302.
16. Xuefei Li, Xiaorong Hu, Jichun Wang. Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway. Int J Mol Med 2018;42(4):1917–24. DOI: 10.3892/ijmm.2018.3794.
17. Perry C.N., Kyoi S., Hariharan N. Novel methods for measuring cardiac autophagy. Methods Enzymol 2009;453:325–42. DOI: 10.1016/S0076-6879(08)04016-0.
18. Maheswari U., Sadras S.R. Mechanism and Regulation of Autophagy in Cancer. Crit Rev Oncog 2018;23(5v6):269–80. DOI: 10.1615/CritRevOncog.2018028394.
19. Wang Z., Liu G., Jiang J. Profiling of apoptosis- and autophagy-associated molecules in human lung cancer A549 cells in response to cisplatin treatment. It J Oncol 2019;543:1071–85. DOI: 10.3892/ijo.2019.4690.
20. Wirawan E. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death & Disease 2010;1(1):e18. DOI: 10.1038/cddis.2009.16.
21. Pasquier B. Autophagy inhibitors. Cell Mol Life Sci 2016;73(5):985–1001. DOI: 10.1007/s00018-015-2104-y.
22. Мерабишвили B.M., Арсеньев А.И., Тарков С.А. Заболеваемость и смертность населения от рака легкого, достоверность учета. Сибирский онкологический журнал 2018;17(6):15–26. DOI.org/10.21294/1814-4861-2018-17-6-15-26.
23. Xu Z., Han X., Ou D., Liu T. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol 2020;104(2):575–87. DOI: 10.1007/s00253-019-10257-8.
24. Noonan J., Zarrer J., Murphy B.M. Targeting Autophagy in Glioblastoma. Crit Rev Oncog 2016;21(3–4):241–52. DOI: 10.1615/CritRevOncog.
25. Sun S.Y., Kurie J.M., Yue P. Differential responses of normal, premalignant, and malignant human bronchial epithelial cells to receptor-selective retinoids. Cancer Res 1999;5(2):431–7. PMID: 10037194.
26. Sun S.Y., Yue P., Chen X. The synthetic retinoid CD437 selectively induces apoptosis in human lung cancer cells while sparing normal human lung epithelial cells.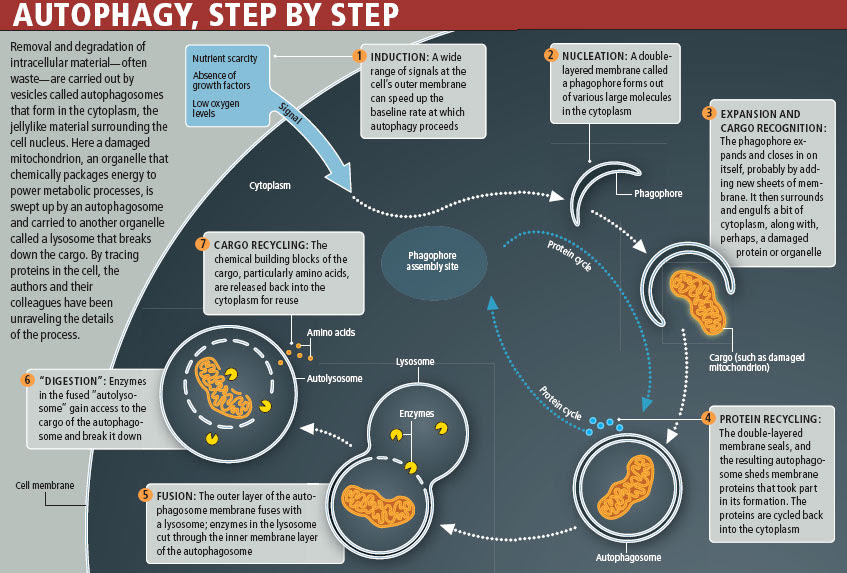 Cancer Res 2002;62(8):2430–6. PMID: 11956107.
Cancer Res 2002;62(8):2430–6. PMID: 11956107.
ВЛИЯНИЕ АУТОФАГИИ НА РЕПЛИКАЦИЮ ВИРУСА КРАСНУХИ | Гулимов
1. Дмитриев Г.В., Борисова Т.К., Файзулоев Е.Б., Забияка Ю.И., Десятскова Р.Г., Зверев В.В. Изучение молекулярных механизмов аттенуации вируса краснухи на примере отечественного штамма С-77. Молекулярная генетика, микробиология и вирусология. 2012, 27(3): 28-34.
2. Adamo M.P., Zapata M., Frey T.K. Analysis of gene expression in fetal and adult cells infected with rubella virus. Virology. 2008, 370(1): 1-11.
3. Chang H., Li X., Cai Q. et al. The PI3K/Akt/mTOR pathway is involved in CVB3-induced autophagy of HeLa cells. International Journal of Molecular Medicine. 2017, 40(1): 182-192.
4. Delgado M., Singh S., De Haro S. et al. Autophagy and pattern recognition receptors in innate immunity. Immunological Reviews. 2009, 227(1): 189-202.
5. Deretic V., Levine B. Autophagy, Immunity, and Microbial Adaptations. Cell Host Microbe. 2009, 5(6): 527-549.
6. Jager S., Bucci C., Tanida I. et al. Role for Rab7 in maturation of late autophagic vacuoles. Journal of Cell Science. 2004, 117(20): 4837-4848.
7. Jordan T.X., Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes and Infection. 2012, 14(2): 126-139.
8. Heaton N.S., Randall G. Dengue Virus-Induced Autophagy Regulates Lipid Metabolism.:max_bytes(150000):strip_icc()/4210008_color4-5c4e3f904cedfd0001ddb4d0.png) Cell Host Microbe. 2010, 8 (5): 422-432.
Cell Host Microbe. 2010, 8 (5): 422-432.
9. Lamark T., Svenning S., Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays in Biochemistry. 2017, 61(6): 609-624.
10. Liang Q., Luo Z., Zeng J. et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem. Cell. 2016, 19 (5): 1-9.
11. Mizushima N., Levine B., Cuervo A.M. et al. Autophagy fights disease through cellular self-digestion. Nature. 2008, 451(7182): 1069-1075.
12. Nazme N.I., Hussain M., Das A.C. Congenital Rubella Syndrome — A Major Review and Update. Delta Med. Col. J. 2015, 3(2): 89-95.
13. Orosz L., Megyeri K. Well begun is half done: Rubella virus perturbs autophagy signaling, thereby facilitating the construction of viral replication compartments. Medical Hypotheses. 2016, 89: 16-20.
14. Pásztor K., Orosz L., Seprényi G. et al. Rubella virus perturbs autophagy. Med. Microbiol. Immunol. 2014, 203(5): 323-331.
15. Shi J., Luo H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim. Biophys. Sin. 2012, 44(5): 375-384.
Необходимое самоедство клетки – аналитический портал ПОЛИТ.РУ
Нобелевская премия по физиологии и медицине 2016 года была присуждена профессору Центра передовых исследований Токийского технологического института и почетному профессору Токийского университета Ёсинори Осуми, объяснившему механизм клеточной аутофагии. Основные работы Осуми, посвященные этой проблеме, были опубликованы в 1990-х годах. В XXI веке ученый уже получил несколько научных премий, включая премию Киото за достижения в науках о жизни (2015).
Основные работы Осуми, посвященные этой проблеме, были опубликованы в 1990-х годах. В XXI веке ученый уже получил несколько научных премий, включая премию Киото за достижения в науках о жизни (2015).
Аутофагия в клетках была обнаружена еще в 1960-е, когда ученые увидели, как клетка переваривает часть собственного содержимого, окружив ее мембранной оболочкой. Получившийся пузырек доставлялся в лизосомы – клеточные органы, служащие для переваривания. Внутри лизосом выделяются гидролитические ферменты, способные расщеплять макромолекулы. В растительных клетках и клетках дрожжей роль лизосом выполняют вакуоли. Бельгийский ученый Кристиан де Дюв (Christian René de Duve) был удостоен Нобелевской премии по физиологии и медицине в 1974 году за открытие лизосом. Он же ввел термин «аутофагия» и назвал пузырьки-везикулы, доставляемые для уничтожения в лизосомы, аутофагосомами.
Принцип клеточной аутофагии
Долгое время было известно лишь, что аутофагосома формируется вокруг подлежащего “разборке” материала, например, поврежденных клеточных органелл, затем путешествует к лизосоме, сливается с ней, и содержимое расщепляется лизосомными ферментами. Лизосому в таком случае часто называют аутолизосомой.
Аутофагосомы (AP) и аутолизосомы (AL) в клетках мухи дрозофилы
В 70-х и 80-х годах основные успехи были достигнуты в изучении другого механизма расщепления белков в клетке – протеолитической деградации (протеолизма). Он возникает благодаря крупным молекулам ферментов – протеасомам. Сначала небольшой белок убиквитин присоединяется к белку, подлежащему расщеплению. На это реагирует протеасома, присоединяется в свою очередь к убиквитину и начинает свою работу. Этот механизм обеспечивает деградацию 80 – 90 % белков в клетке. За открытие убиквитин-опосредованной деградации белка в 2004 году Нобелевскую премию по химии получили Аарон Чехановер, Аврам Гершко и Ирвин Роуз.
Хоть протеасомы и выполняют большую часть работы по уничтожению белков, меньшая по объему, но не меньшая по важности работа приходится на механизм лизосомной аутофагии.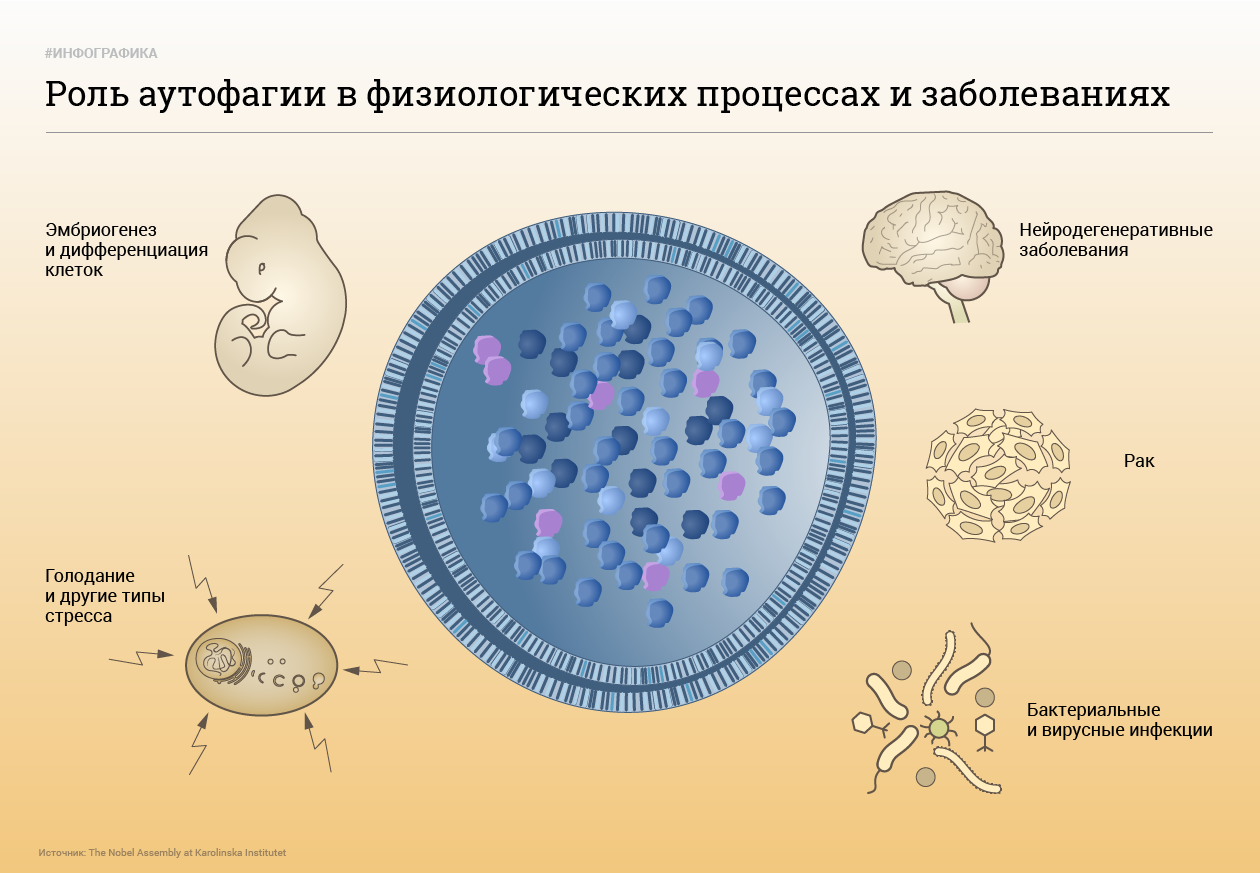 Именно он позволяет клетке избавляться от крупных белковых комплексов и изношенных органелл. В своей лаборатории Ёсинори Осуми провел ряд экспериментов с обычными пекарскими дрожжами (Saccharomyces cerevisiae), благодаря которым выяснил, какие гены ответственны за аутофагию.
Именно он позволяет клетке избавляться от крупных белковых комплексов и изношенных органелл. В своей лаборатории Ёсинори Осуми провел ряд экспериментов с обычными пекарскими дрожжами (Saccharomyces cerevisiae), благодаря которым выяснил, какие гены ответственны за аутофагию.
Правда, в начале ученому пришлось решить одну проблему. Клетки дрожжей малы, и в то время они не были достаточно изучены, так что у биологов даже не было уверенность, что в вакуолях дрожжевых клеток происходит тот же процесс аутофагии, что и в лизосомах животных. Разглядеть этот процесс под микроскопом не удавалось. Осуми рассудил, что доказательством послужит накопление аутофагосом в вакуолях дрожжевых клеткок, лишенных расщепляющих белки ферментов. Он получил мутантные клетки без этих ферментов, и действительно их вакуоли стали довольно быстро наполняться множеством мелких пузырьков.
Экспериментируя с геномом дрожжевых клеток, вызывая в нем различные мутации, Осуми сумел установить 15 генов, связанных с механизмом аутофагии. Затем он перешел к исследованию контролируемых этими клетками белков. Результаты показали, что весь процесс аутофагии управляется каскадом белков и белковых комплексов, каждый из которых регулирует отдельную стадию формирования аутофагосом. Работы Осуми не только выявили механизм аутофагии у дрожжевых клеток, но и дали ученым исследовательские методы, позволившие изучать аутофагию уже и в клетках человека. Благодаря его открытию, стала ясной фундаментальная роль аутофагии в жизни организма.
“Сферы применения” аутофагии
Сейчас ученым уже известно, что аутофагия выполняет целый ряд функций. Она помогает быстро обеспечить клетку питательными веществами, поэтому включается в механизм клеточного ответа на голод и другие виды стресса. Эксперименты на дрожжах показали, что при голодании уровень аутофагии повышается. Это позволяет организму, расщепив ненужные белки на аминокислоты, использовать эти аминокислоты для синтеза белков, необходимых для выживания в первую очередь. Такое же явление обнаружено и на лабораторных мышах. Обнаружено, что у млекопитающих аутофагия активизируется сразу после рождения, когда организм перестает снабжаться питанием через плаценту.
Такое же явление обнаружено и на лабораторных мышах. Обнаружено, что у млекопитающих аутофагия активизируется сразу после рождения, когда организм перестает снабжаться питанием через плаценту.
Помеченные флуоресцентной меткой аутофагосомы в клетках печени голодающей мыши
Механизм аутофагии участвует и в уничтожении проникших в клетку инфекционных частиц (в данном случае это уже не ауто-, а ксенофагия), поэтому она важна для нормального функционирования врожденного иммунитета. Например, Mycobacterium tuberculosis, вызывающие туберкулез, уничтожаются в клетках точно так же, как отработавшие свой срок митохондрии (кстати, ученые считают этот факт еще одним доказательством, что митохондрии до того, как превратиться в клеточный орган, были самостоятельными бактериями). Некоторые болезнетворные бактерии приобретают способность блокировать процесс своего переваривания, поэтому стимуляция аутофагии может стать в этих случаях методом лечения.
Аутофагия позволяет клетке распознавать вирус везикулярного стоматита (Indiana vesiculovirus) и запускать противовирусный иммунный ответ. Вирус захватывается аутофагосомой и доставляется в эндосому, где его распознает толл-подобный рецептор 7. Рецептор реагирует на одноцепочечную молекулу РНК вируса. После детекции вируса рецептор запускает сигнальный каскады, который приводит к выработке интерферона и других противовирусных цитокинов.
Поскольку именно аутофагия нужна для избавления от поврежденных органелл, клеточных мембран и белков, нарушение ее механизма оказывается одной из главных причин накопления повреждений, а значит и старения клетки. Вероятно, с аутофагией связан и один из механизмов программируемой клеточной смерти. Однако аутофагическая активность в отмирающих клетках может быть как причиной их смерти, так и попыткой выжить. Исследования в данной области продолжаются. Их проводят, например, на насекомых, у которых в процессе превращения из личинки в куколку и во взрослую особь задействованы механизмы программируемой клеточной смерти. Некоторые результаты показывают, что тип регуляции аутофагии, направленной на выживания и на гибель клетки, отличаются.
Аутофагия задействована также на стадии эмбриогенеза при дифференциации разных типов клеток. Нарушения механизма аутофагии связаны с возникновением рака и некоторых других заболеваний, например, они отмечаются при болезни Паркинсона. Сейчас ученые активно работают над созданием лекарств, которые регулировали бы именно процесс аутофагии.
Аутофагосома — Википедия
Share
Pin
Tweet
Send
Share
Send
An аутофагосома представляет собой сферическую структуру с двухслойными мембранами. Это ключевая структура в макроавтофагия, система внутриклеточной деградации цитоплазматического содержимого (например, аномальные внутриклеточные белки, избыточные или поврежденные органеллы, вторгающиеся микроорганизмы). После образования аутофагосомы доставляют цитоплазматические компоненты к лизосомы. Наружная мембрана аутофагосомы сливается с лизосомой, образуя автолизосома. Гидролазы лизосомы разрушают доставляемое аутофагосомами содержимое и его внутреннюю мембрану.[1]
Образование аутофагосом регулируется генами, которые хорошо законсервированы от дрожжей до высших эукариот. Номенклатура этих генов различалась от бумаги к бумаге, но в последние годы она была упрощена. Семейства генов, ранее известные как APG, AUT, CVT, GSA, PAZ и PDD, теперь объединены в семейство ATG (родственное AuTophaGy).[2]
Размер аутофагосом варьируется от млекопитающие и дрожжи. Аутофагосомы дрожжей имеют размер около 500-900 нм, а аутофагосомы млекопитающих больше (500-1500 нм). В некоторых примерах ячеек, например эмбриональные стволовые клетки, эмбриональные фибробласты и гепатоциты, аутофагосомы видны при световой микроскопии и могут рассматриваться как кольцеобразные структуры.[1]
Формирование аутофагосом
Начальный этап формирования аутофагосом омегазом на эндоплазматический ретикулумс последующим удлинением структур, называемых фагофорами. [3]
[3]
Формирование аутофагосом контролируется генами Atg через комплексы Atg12-Atg5 и LC3. Конъюгат Atg12-Atg5 также взаимодействует с Atg16 с образованием более крупных комплексов. Модификация Atg5 с помощью Atg12 необходима для удлинения исходной мембраны.[4]
После образования сферической структуры комплекс ATG12-ATG5:ATG16L1 отделяется от аутофагосомы. LC3 раскалывается ATG4 протеаза для генерации цитозольного LC3. Расщепление LC3 необходимо для терминального слияния аутофагосомы с ее мембраной-мишенью. LC3 обычно используется в качестве маркера аутофагосом в иммуноцитохимия, потому что это основная часть пузырька и остается связанным до последнего момента перед слиянием. Сначала аутофагосомы сливаются с эндосомы или везикулы, происходящие из эндосом. Эти структуры затем называют амфисомами или промежуточными аутофагическими вакуолями.[5] Тем не менее, эти структуры содержат эндоцитарные маркеры, даже небольшие лизосомальные белки, такие как катепсин D.
Этот процесс аналогичен у дрожжей, однако названия генов различаются. Например, LC3 у млекопитающих Atg8 в дрожжах и аутофагосомах образуются из преаутофагосомной структуры (PAS), которая отличается от структур-предшественников в клетках млекопитающих. Преаутофагосомная структура у дрожжей описывается как комплекс, локализованный около вакуоли. Однако значение этой локализации неизвестно. Зрелые аутофагосомы дрожжей сливаются непосредственно с вакуолями или лизосомами и не образуют амфисомы, как у млекопитающих.[6]
В созревании аутофагосом дрожжей есть также другие известные участники, как Atg1, Atg13 и Atg17. Atg1 представляет собой киназу, активируемую при индукции аутофагии. Atg13 регулирует Atg1, и вместе они образуют комплекс под названием Atg13: Atg1, который получает сигналы от мастера восприятия питательных веществ — Tor. Atg1 также важен на поздних стадиях формирования аутофагосом.[6]
Функция в нейронах
В нейроны, аутофагосомы образуются в нейрит кончиках и созревают (подкисляются) по мере продвижения к телу клетки по аксон.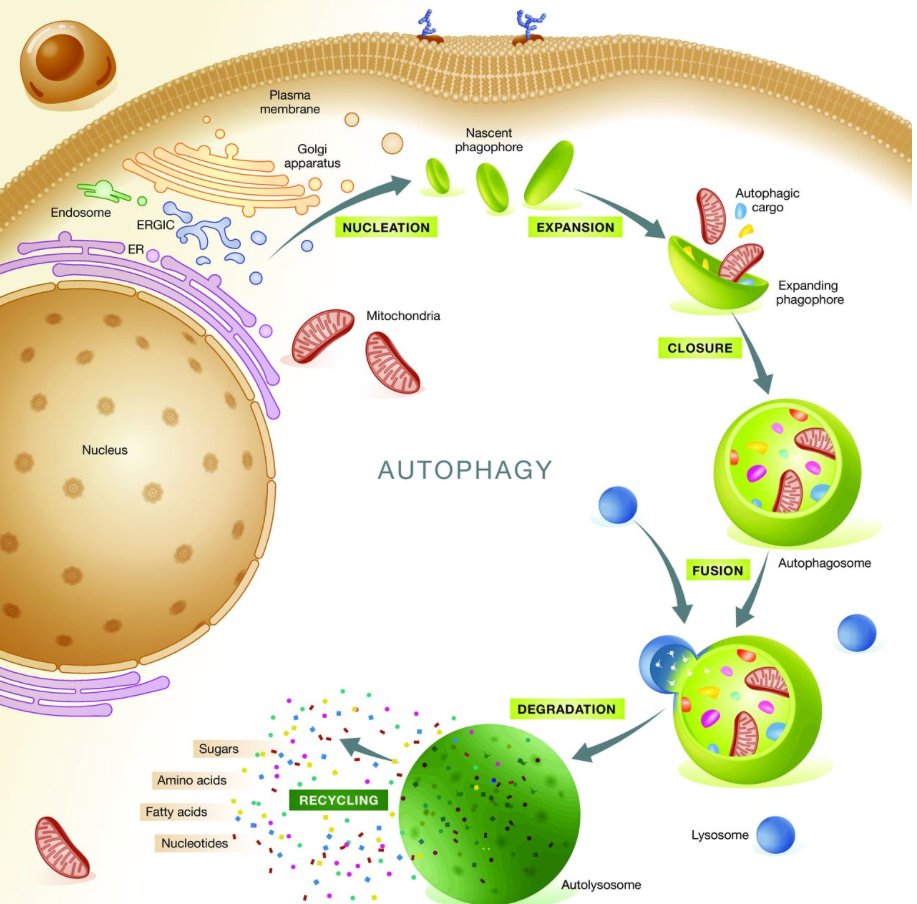 Wong, Y.C .; Хольцбаур, Э. Л. (2014). «Регулирование динамики аутофагосом с помощью хантинтина и HAP1 нарушается экспрессией мутантного хантинтина, что приводит к деградации дефектных грузов». Журнал неврологии. 34 (4): 1293–305. Дои:10.1523 / JNEUROSCI.1870-13.2014. ЧВК 3898289. PMID 24453320.
Wong, Y.C .; Хольцбаур, Э. Л. (2014). «Регулирование динамики аутофагосом с помощью хантинтина и HAP1 нарушается экспрессией мутантного хантинтина, что приводит к деградации дефектных грузов». Журнал неврологии. 34 (4): 1293–305. Дои:10.1523 / JNEUROSCI.1870-13.2014. ЧВК 3898289. PMID 24453320.
Share
Pin
Tweet
Send
Share
Send
Новые сведения о слиянии аутофагосомы и лизосомы | Журнал клеточной науки
Макроаутофагия, далее именуемая аутофагией, представляет собой катаболический процесс, нацеленный на широкий спектр клеточных компонентов, включая белки, липиды, поврежденные органеллы и патогены. Аутофагия обычно происходит на базальном уровне, но она ускоряется различными стрессами, такими как голод, накопление аномальных белков, повреждение органелл и инфекция патогенами.Первоначально аутофагия считалась системой массового неселективного разложения; однако теперь известно, что аутофагия избирательно разрушает мишени и способствует внутриклеточному гомеостазу (Kawabata and Yoshimori, 2016). Во время аутофагии небольшая цистерна, называемая изоляционной мембраной (фагофор), удлиняется и окружает часть цитоплазмы, образуя двухмембранную структуру, называемую аутофагосомой. Аутофагосомы либо сливаются с поздними эндосомами с образованием амфисом, которые затем сливаются с лизосомами, либо они сливаются непосредственно с лизосомами (Berg et al., 1998; Fader et al., 2008). После слияния с лизосомами они называются автолизосомами, а изолированное содержимое переваривается (рис. 1).
Рис. 1.
Обзор аутофагии. При индукции аутофагии стрессом цитоплазматический материал изолируется двухмембранной структурой, называемой аутофагосомой. Эти аутофагосомы сливаются с поздними эндосомами (называемыми амфисомами) или лизосомами, чтобы стать автолизосомами, в которых секвестрированные грузы разлагаются и рециркулируются для поддержания клеточного гомеостаза. Аутофагию можно разделить на несколько этапов: формирование изолирующей мембраны (нуклеация), удлинение изолирующей мембраны (удлинение), завершение и транспорт аутофагосомы (созревание), стыковка и слияние аутофагосомы и лизосомы (слияние) и деградация грузы внутри автолизосомы (деградация).
Аутофагию можно разделить на несколько этапов: формирование изолирующей мембраны (нуклеация), удлинение изолирующей мембраны (удлинение), завершение и транспорт аутофагосомы (созревание), стыковка и слияние аутофагосомы и лизосомы (слияние) и деградация грузы внутри автолизосомы (деградация).
Рис. 1.
Обзор аутофагии. При индукции аутофагии стрессом цитоплазматический материал изолируется двухмембранной структурой, называемой аутофагосомой.Эти аутофагосомы сливаются с поздними эндосомами (называемыми амфисомами) или лизосомами, чтобы стать автолизосомами, в которых секвестрированные грузы разлагаются и рециркулируются для поддержания клеточного гомеостаза. Аутофагию можно разделить на несколько этапов: формирование изолирующей мембраны (нуклеация), удлинение изолирующей мембраны (удлинение), завершение и транспорт аутофагосомы (созревание), стыковка и слияние аутофагосомы и лизосомы (слияние) и деградация грузы внутри автолизосомы (деградация).
После идентификации генов, связанных с аутофагией (ATGs) у дрожжей (Tsukada and Ohsumi, 1993), функции их гомологов были идентифицированы и широко изучены, особенно в клетках мышей и млекопитающих (Mizushima et al., 2011). Вкратце, активация комплекса unc-51-like kinase 1 (ULK1; Atg1 у дрожжей) имеет решающее значение для инициации аутофагии. Затем активация комплекса фосфатидилинозитол-3-киназы класса III, который включает PI3K (Vps34 в дрожжах), беклин 1, VPS15 (PIK3R4) и ATG14L (ATG14), запускает зарождение везикул.Последующее удлинение и закрытие изолирующей мембраны опосредуется двумя убиквитин-подобными путями конъюгации ATG: ATG5-ATG12 и LC3 / Atg8. У млекопитающих существует семь ортологов Atg8; MAP1LC3A / B / C, GABARAP и GABARAPL1 / 2/3 (все из которых далее именуются LC3). LC3 широко используется в качестве маркера для микроскопического обнаружения изолирующих мембран и аутофагосом. После синтеза LC3 обрабатывается на своем С-конце с помощью Atg4 и превращается в LC3-I, который имеет остаток глицина на С-конце.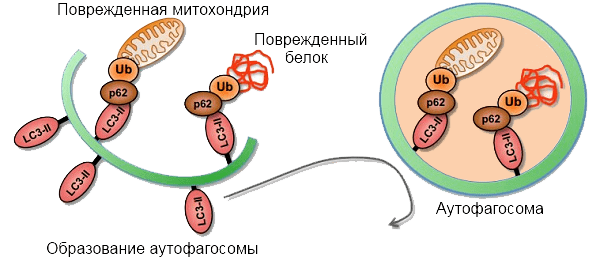 LC3-I впоследствии конъюгируется с фосфатидилэтаноламином (PE), чтобы стать LC3-II посредством ферментативной реакции, подобной убиквитинированию. В отличие от цитоплазматической локализации LC3-I, LC3-II ассоциирует как с внешней, так и с внутренней мембранами аутофагосомы. PE-конъюгированный LC3 (LC3-II) и неконъюгированный LC3 (LC3-I) могут быть обнаружены отдельно с помощью иммуноблот-анализа, а количество LC3-II также широко используется для количественной оценки аутофагической активности (Kabeya et al., 2000) .
LC3-I впоследствии конъюгируется с фосфатидилэтаноламином (PE), чтобы стать LC3-II посредством ферментативной реакции, подобной убиквитинированию. В отличие от цитоплазматической локализации LC3-I, LC3-II ассоциирует как с внешней, так и с внутренней мембранами аутофагосомы. PE-конъюгированный LC3 (LC3-II) и неконъюгированный LC3 (LC3-I) могут быть обнаружены отдельно с помощью иммуноблот-анализа, а количество LC3-II также широко используется для количественной оценки аутофагической активности (Kabeya et al., 2000) .
Хотя обширная характеристика генов ATG дала понимание механизмов активации аутофагии и образования аутофагосом, то, как контролируется слияние аутофагосом с эндосомами и / или лизосомами, остается малоизученным.Тем не менее, недавние исследования начали раскрывать молекулярные механизмы, которые регулируют стадии слияния. Несколько экспериментальных подходов способствовали идентификации условий, которые необходимы для того, чтобы произошла стадия слияния аутофагосома-лизосома. Например, ингибитор V-АТФазы бафиломицин A1 (BafA1), макролидный антибиотик, полученный из Streptomyces griseus , который блокирует деградацию в аутолизосомах и / или слияние аутофагосома-лизосома (Klionsky et al., 2008; Yamamoto et al., 1998; Yoshimori et al., 1991) и тандем LC3, помеченный RFP и GFP (RFP-GFP-LC3), который теряет свою флуоресценцию GFP после слияния с лизосомой, помогли обнаружить блокировку слияния аутофагосом (Kimura et al., 2007). В этом комментарии мы рассматриваем недавние открытия в отношении молекулярных механизмов, лежащих в основе стадии слияния аутофагосома-лизосома, с акцентом на исследованиях на млекопитающих, а также обсуждаем будущие перспективы в этой области.
Время слияния аутофагосома-лизосома очень важно, и только закрытые аутофагосомы могут сливаться с лизосомами. Это поднимает вопрос, как регулируется закрытие аутофагосом. У млекопитающих дефект в системе конъюгации ATG приводит к накоплению незамкнутых аутофагосом (Fujita et al., 2008a; Kishi-Itakura et al., 2014; Mizushima et al., 2001; Sou et al., 2008), что подразумевает что он, вероятно, действует при удлинении и закрытии аутофагосом и важен для перехода изолирующей мембраны в аутофагосому. Помимо своей роли в созревании аутофагосом, недавно было показано, что система конъюгации ATG (состоящая из ATG3, ATG5 и ATG7) необходима для эффективного разрушения внутренней аутофагической мембраны; однако это не требуется для слияния аутофагосома-лизосома, хотя скорость образования аутофагосом снижается до ~ 30% в клетках с дефицитом конъюгации ATG (Tsuboyama et al., 2016). Напротив, другое исследование, в котором использовались клеточные линии, в которых было отключено все семейство белков ATG8, показало, что белки LC3 и GABARAP не требуются для образования аутофагосом, но имеют решающее значение для слияния аутофагосома-лизосома (Nguyen et al., 2016) . Отсутствие слияния, вероятно, связано с нарушением рекрутирования адапторного белка PLEKHM1 (McEwan et al., 2015) (см. Также ниже) в аутофагосомы. Несоответствие между этими двумя исследованиями может отражать неперекрывающуюся функцию системы конъюгации ATG и LC3 и GABARAP.Почему только закрытые аутофагосомы распознаются несколькими факторами слияния (обсуждаемыми ниже), в настоящее время неясно.
Это поднимает вопрос, как регулируется закрытие аутофагосом. У млекопитающих дефект в системе конъюгации ATG приводит к накоплению незамкнутых аутофагосом (Fujita et al., 2008a; Kishi-Itakura et al., 2014; Mizushima et al., 2001; Sou et al., 2008), что подразумевает что он, вероятно, действует при удлинении и закрытии аутофагосом и важен для перехода изолирующей мембраны в аутофагосому. Помимо своей роли в созревании аутофагосом, недавно было показано, что система конъюгации ATG (состоящая из ATG3, ATG5 и ATG7) необходима для эффективного разрушения внутренней аутофагической мембраны; однако это не требуется для слияния аутофагосома-лизосома, хотя скорость образования аутофагосом снижается до ~ 30% в клетках с дефицитом конъюгации ATG (Tsuboyama et al., 2016). Напротив, другое исследование, в котором использовались клеточные линии, в которых было отключено все семейство белков ATG8, показало, что белки LC3 и GABARAP не требуются для образования аутофагосом, но имеют решающее значение для слияния аутофагосома-лизосома (Nguyen et al., 2016) . Отсутствие слияния, вероятно, связано с нарушением рекрутирования адапторного белка PLEKHM1 (McEwan et al., 2015) (см. Также ниже) в аутофагосомы. Несоответствие между этими двумя исследованиями может отражать неперекрывающуюся функцию системы конъюгации ATG и LC3 и GABARAP.Почему только закрытые аутофагосомы распознаются несколькими факторами слияния (обсуждаемыми ниже), в настоящее время неясно.
Цитоскелет выполняет множество функций, включая структурное поддержание клеток, деление и движение клеток. Хотя микротрубочки незаменимы для аутофагии у дрожжей (Kirisako et al., 1999), они необходимы для стадии слияния у млекопитающих (Aplin et al., 1992; Kochl et al., 2006; Монастырская и др., 2009). Считается, что аутофагосомы образуются случайным образом по всей цитоплазме, тогда как поздние эндосомы и лизосомы преимущественно находятся в перинуклеарной области. Следовательно, как только были созданы полные и закрытые аутофагосомы, их необходимо доставить в перинуклеарную область. Направленный на минус-конец моторный комплекс динеин-динактин перемещает груз в перинуклеарную область, тогда как большинство кинезинов представляют собой двигательные белки, направленные на положительный конец, которые направляют свой груз к периферии клетки (Gross et al., 2007). Учитывая, что лизосомы локализуются в перинуклеарных регионах, направленный на минус-конец транспорт аутофагосом кажется разумным и, действительно, визуализация в реальном времени показывает, что зрелые аутофагосомы движутся по дорожкам микротрубочек к лизосомам (Kimura et al., 2008). Эффективное движение аутофагосом ингибируется микроинъекцией антител против LC3, что указывает на роль LC3 в этом процессе (Kimura et al., 2008). Кроме того, кинезин-зависимый транспорт, направленный на плюс-конец, важен для правильного позиционирования аутофагосом, поскольку истощение кинезина KIF5B блокирует аутофагию и приводит к перинуклеарному кластеризации аутофагосом (Cardoso et al., 2009). Подобно млекопитающим, у Drosophila PX-домен-содержащий кинезин Klp98A контролирует образование, слияние и внутриклеточное позиционирование аутофагических пузырьков (Mauvezin et al., 2016).
Следовательно, как только были созданы полные и закрытые аутофагосомы, их необходимо доставить в перинуклеарную область. Направленный на минус-конец моторный комплекс динеин-динактин перемещает груз в перинуклеарную область, тогда как большинство кинезинов представляют собой двигательные белки, направленные на положительный конец, которые направляют свой груз к периферии клетки (Gross et al., 2007). Учитывая, что лизосомы локализуются в перинуклеарных регионах, направленный на минус-конец транспорт аутофагосом кажется разумным и, действительно, визуализация в реальном времени показывает, что зрелые аутофагосомы движутся по дорожкам микротрубочек к лизосомам (Kimura et al., 2008). Эффективное движение аутофагосом ингибируется микроинъекцией антител против LC3, что указывает на роль LC3 в этом процессе (Kimura et al., 2008). Кроме того, кинезин-зависимый транспорт, направленный на плюс-конец, важен для правильного позиционирования аутофагосом, поскольку истощение кинезина KIF5B блокирует аутофагию и приводит к перинуклеарному кластеризации аутофагосом (Cardoso et al., 2009). Подобно млекопитающим, у Drosophila PX-домен-содержащий кинезин Klp98A контролирует образование, слияние и внутриклеточное позиционирование аутофагических пузырьков (Mauvezin et al., 2016).
Интересно, что локализация лизосом определяет скорость слияния аутофагосом. Увеличение перинуклеарной локализации лизосом за счет истощения кинезинов KIF1B-β и KIF2A приводит к усилению слияния аутофагосом, тогда как распространение лизосом на периферию за счет сверхэкспрессии моторов снижает скорость слияния (Корольчук и др., 2011). Таким образом, скоординированный транспорт как аутофагосом, так и лизосом важен для слияния; но как аутофагосомы и лизосомы связаны с микротрубочками? Малая GTPase Rab7, которая действует как молекулярный переключатель и, предположительно, рекрутируется в поздние аутофагосомы (Gutierrez et al.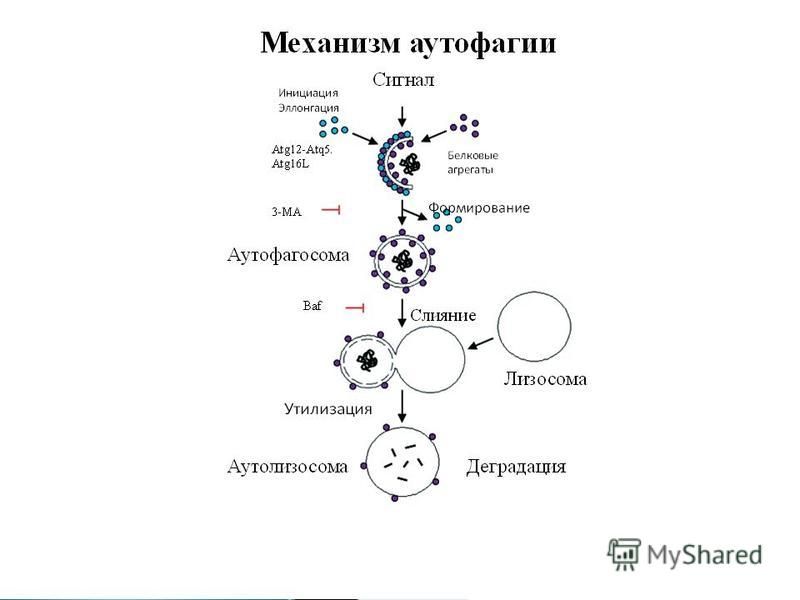 , 2004), связывает аутофагосомы с двигателями микротрубочек через FYCO1 (FYVE и домен, содержащий спиральную спираль 1), тем самым опосредуя кинезин-управляемое движение к периферии клетки (см. ниже) (Pankiv et al., 2010). Rab7 также работает в обратном направлении, взаимодействуя с Rab-взаимодействующим лизосомным белком (RILP), датчиком холестерина ORP1L (также известным как OSBPL1A) и динеином, чтобы облегчить транспорт аутофагосом, автолизосом и лизосом в перинуклеарную область (Jordens et al. al., 2001; Wijdeven et al., 2016) (рис.2).
, 2004), связывает аутофагосомы с двигателями микротрубочек через FYCO1 (FYVE и домен, содержащий спиральную спираль 1), тем самым опосредуя кинезин-управляемое движение к периферии клетки (см. ниже) (Pankiv et al., 2010). Rab7 также работает в обратном направлении, взаимодействуя с Rab-взаимодействующим лизосомным белком (RILP), датчиком холестерина ORP1L (также известным как OSBPL1A) и динеином, чтобы облегчить транспорт аутофагосом, автолизосом и лизосом в перинуклеарную область (Jordens et al. al., 2001; Wijdeven et al., 2016) (рис.2).
Рис. 2.
Транспорт аутофагосом. Rab7 GTPase связывает аутофагосомы с двигателем микротрубочек через FYCO1, чтобы облегчить управляемое кинезином движение к периферии клетки.Было установлено, что FYCO1 связывается с LC3, но он также связывается с фосфолипидом PI3P, компонентом мембраны аутофагосомы. Rab7 также связывается с RILP и ORP1L, чтобы опосредовать динеин и / или управляемое динактином движение к перинуклеарной области при нормальных условиях холестерина. При низком уровне холестерина ORP1L образует сайт контакта с VAP-A, который предотвращает рекрутирование динактина и блокирует транспорт минус-конца (не показан на этом рисунке). Позиционирование лизосом также определяет скорость слияния аутофагосома-лизосома.
Рис. 2.
Транспорт аутофагосом. Rab7 GTPase связывает аутофагосомы с двигателем микротрубочек через FYCO1, чтобы облегчить управляемое кинезином движение к периферии клетки. Было установлено, что FYCO1 связывается с LC3, но он также связывается с фосфолипидом PI3P, компонентом мембраны аутофагосомы. Rab7 также связывается с RILP и ORP1L, чтобы опосредовать динеин и / или управляемое динактином движение к перинуклеарной области при нормальных условиях холестерина.При низком уровне холестерина ORP1L образует сайт контакта с VAP-A, который предотвращает рекрутирование динактина и блокирует транспорт минус-конца (не показан на этом рисунке). Позиционирование лизосом также определяет скорость слияния аутофагосома-лизосома.
Позиционирование лизосом также определяет скорость слияния аутофагосома-лизосома.
Подобно микротрубочкам, актиновые филаменты образуют дорожки для перемещения различных внутриклеточных грузов с помощью семейства моторных белков миозина. Некоторые доказательства указывают на то, что актин участвует в слиянии аутофагосом и лизосом.Напр., Гистондеацетилаза 6 (HDAC6) задействует зависимый от кортактина аппарат ремоделирования актина, который в свою очередь собирает актиновую сеть, которая стимулирует слияние аутофагосома-лизосома (Lee et al., 2010). Интересно, что HDAC6 и сборка актина незаменимы для вызванной голоданием аутофагии, но необходимы для избирательной деградации агрегированных белков (Lee et al., 2010). Более того, актин моторный миозин VI и Tom1, компонент механизма ESCRT и партнер связывания миозина V1 на эндосомах, участвуют в слиянии, поскольку потеря обоих факторов снижает аутофагосомальную доставку эндоцитарного груза и блокирует слияние аутофагосома-лизосома (Tumbarello и другие., 2012).
Малые GTPases семейства Ras-родственного белка мозга (Rab) являются эволюционно законсервированными и важными регуляторами мембранного переноса в эукариотических клетках. Они привлекают специфические эффекторные белки, такие как адаптеры груза, для формирования транспортных везикул, моторные белки для перемещения везикулы к своей целевой мембране, а также связывающие белки, чтобы помочь механизму слияния, когда везикулярный груз достигает места назначения (Stenmark, 2009; Zhen and Стенмарк, 2015).Каждый Rab-белок локализуется в отдельном мембранном компартменте, и, таким образом, Rabs, как полагают, обеспечивают специфичность мембранного транспорта. Связанные с мембраной Rab активируются специфическими факторами обмена гуаниновых нуклеотидов (GEF), которые управляют связыванием GTP.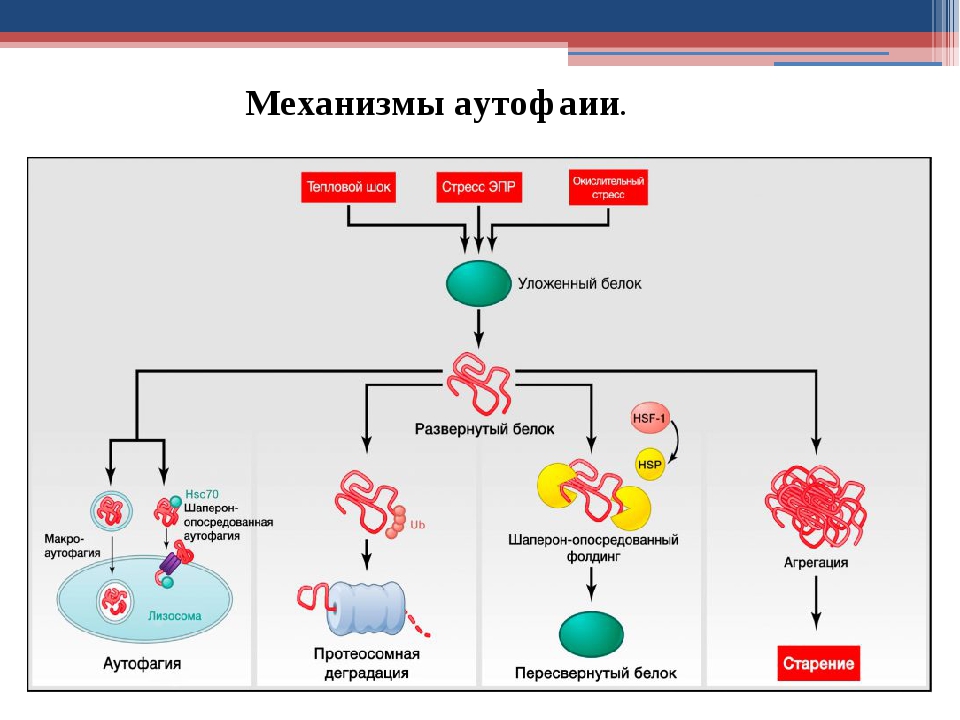 После связывания GTP, Rabs конформационно изменяются, чтобы взаимодействовать со своими эффекторными белками. Впоследствии Rab инактивируются специфическими белками, активирующими GTPase (GAP), которые гидролизуют связанный GTP до GDP, вызывая потерю эффекторного связывания и извлечение из мембран.
После связывания GTP, Rabs конформационно изменяются, чтобы взаимодействовать со своими эффекторными белками. Впоследствии Rab инактивируются специфическими белками, активирующими GTPase (GAP), которые гидролизуют связанный GTP до GDP, вызывая потерю эффекторного связывания и извлечение из мембран.
Было высказано предположение, что некоторые члены семейства Раб регулируют аутофагию. Rab7, который локализован на поздних эндосомах и лизосомах и необходим для последующего переноса эндоцитарной мембраны от поздних эндосом к лизосомам, также важен для слияния аутофагосома-лизосома и последующей деградации содержимого аутофагосомы (Gutierrez et al., 2004; Jager et al., др., 2004; Кирисако и др., 1999). Rab7 также может быть привлечен к поздним аутофагосомам.Gutierrez et al. показали, что при индукции аутофагии наблюдается увеличение интенсивности мечения Rab7 на аутофагической вакуоли (обратите внимание, что термин аутофагическая вакуоль относится к возникающим аутофагосомам и аутофагосомам, которые слились с поздними эндосомами и лизосомами), потому что окрашивание Rab7 на поздних аутофагических вакуоль сильнее, чем на ранних аутофагических вакуолях (по данным иммунофлуоресцентной микроскопии и иммуноэлектронной микроскопии) (Gutierrez et al., 2004). Группа также заявила, что доставка Rab7 в аутофагосомы обнаруживается до слияния с LAMP-1-положительным компартментом.Нокдаун Rab7 вызывает накопление поздних аутофагических вакуолей, указывая тем самым, что функция Rab7 необходима только для окончательного созревания поздних аутофагических вакуолей, вероятно, для слияния с лизосомами (Gutierrez et al., 2004; Jager et al., 2004). Интересно, что Rab7 участвует в формировании GAS-содержащих аутофагосомоподобных структур (GcAVs), которые секвестрируют вторгающиеся стрептококки группы A (Yamaguchi et al., 2009). Rab7 также участвует в расширении изолированной мембраны во время митофагии (Yamano et al. , 2014). Эти результаты показывают, что Rab7 функционирует во время ранней фазы селективных типов аутофагии. Из-за таких множественных и перекрывающихся ролей оказалось трудно определить специфическую роль Rabs во время слияния аутофагосома-лизосома. Тем не менее, тапсигаргин, стрессор ER, широко используемый для индукции аутофагии, блокирует рекрутирование Rab7 в зрелые аутофагосомы и ингибирует их слияние с эндоцитарными пузырьками, не влияя на эндоцитоз, подчеркивая жизненно важную роль Rab7 на этапе слияния (Ganley et al., 2011).
, 2014). Эти результаты показывают, что Rab7 функционирует во время ранней фазы селективных типов аутофагии. Из-за таких множественных и перекрывающихся ролей оказалось трудно определить специфическую роль Rabs во время слияния аутофагосома-лизосома. Тем не менее, тапсигаргин, стрессор ER, широко используемый для индукции аутофагии, блокирует рекрутирование Rab7 в зрелые аутофагосомы и ингибирует их слияние с эндоцитарными пузырьками, не влияя на эндоцитоз, подчеркивая жизненно важную роль Rab7 на этапе слияния (Ganley et al., 2011).
У млекопитающих GEF, который активирует Rab7 для слияния, не идентифицирован. В жировых клетках Drosophila гуанозиновый обменный комплекс Ccz1 и Mon1 (Ccz1-Mon1) рекрутирует Rab7 в PI3P-положительные аутофагосомы (Hegedus et al., 2016), а потеря комплекса Ccz1-Mon1-Rab7 нарушает аутофагосому-лизосому слияние. Здесь Rab5 рекрутирует Ccz1-Mon1 в эндосомы для активации Rab7, что способствует созреванию эндосом и слиянию с лизосомами.Однако Rab5-нулевые мутанты обнаруживают нормальное слияние аутофагосома-лизосома, и Rab5 необязателен для Ccz1-Mon1-зависимого рекрутирования Rab7 (Hegedus et al., 2016).
Два компонента комплекса PI3K, резистентность к УФ-излучению (UVRAG) и Rubicon (RUBCN), участвуют в эндоцитарном транспорте, созревании аутофагосом и / или слиянии аутофагосома-лизосома через Rab7 (Liang et al., 2008; Matsunaga et al., 2009; Табата и др., 2010; Zhong et al., 2009), хотя они имеют противоположные эффекты. UVRAG способствует слиянию аутофагосома-лизосома, тогда как Rubicon ингибирует его. UVRAG, который локализуется в эндоплазматическом ретикулуме и эндосомах, связывается с VPS16, субъединицей комплекса гомотипического слияния и сортировки белков (HOPS) (Liang et al., 2008), чтобы стимулировать активность ГТФазы Rab7 и слияние аутофагосома-лизосома, тогда как Rubicon связывает к UVRAG и отрицательно регулирует активность VPS34 (Sun et al.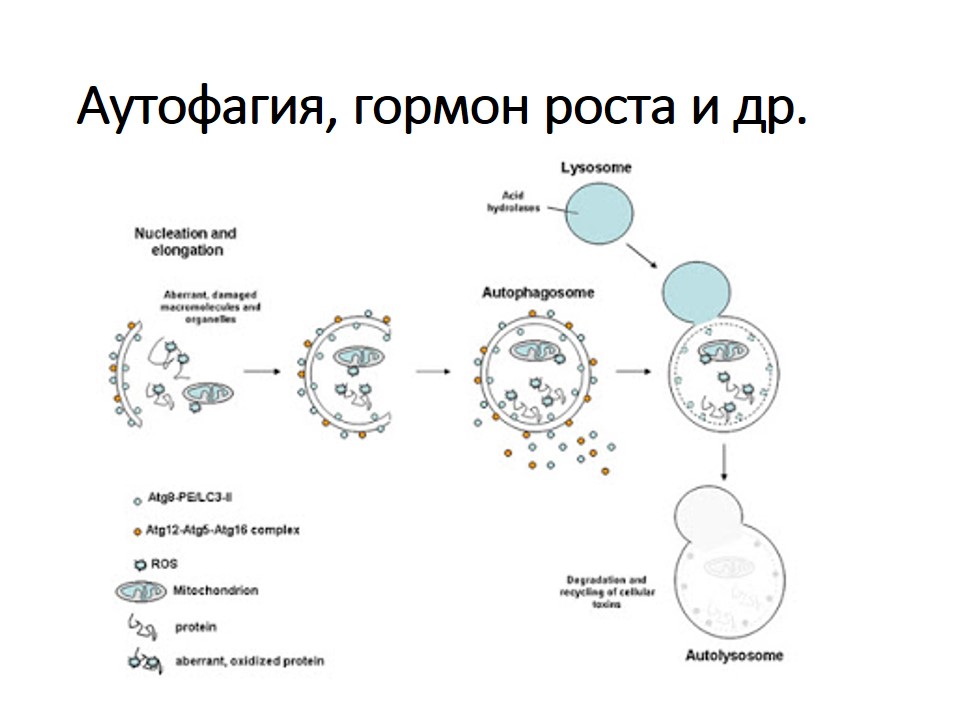 , 2011). В условиях, богатых питательными веществами, UVRAG фосфорилируется механистической мишенью комплекса рапамцицина 1 (mTORC1) (Kim et al., 2015), который усиливает взаимодействие с Rubicon и снижает активность киназы VPS34, а также взаимодействие между UVRAG и комплексом HOPS, тем самым влияя на созревание аутофагосом. Предотвращение фосфорилирования UVRAG увеличивает скорость созревания аутофагосом и лизосомной деградации, указывая на то, что mTORC1 не только регулирует индукцию аутофагии, но также способствует слиянию посредством UVRAG. Однако функция UVRAG во время слияния спорна, потому что другое исследование показало, что UVRAG не взаимодействует с HOPS и не регулирует слияние аутофагосома-лизосома (Jiang et al., 2014). Сходным образом UVRAG незаменим для слияния аутофагосом и лизосом у Drosophila (Takats et al., 2014). Необходимы дальнейшие исследования для уточнения функции UVRAG во время слияния.
, 2011). В условиях, богатых питательными веществами, UVRAG фосфорилируется механистической мишенью комплекса рапамцицина 1 (mTORC1) (Kim et al., 2015), который усиливает взаимодействие с Rubicon и снижает активность киназы VPS34, а также взаимодействие между UVRAG и комплексом HOPS, тем самым влияя на созревание аутофагосом. Предотвращение фосфорилирования UVRAG увеличивает скорость созревания аутофагосом и лизосомной деградации, указывая на то, что mTORC1 не только регулирует индукцию аутофагии, но также способствует слиянию посредством UVRAG. Однако функция UVRAG во время слияния спорна, потому что другое исследование показало, что UVRAG не взаимодействует с HOPS и не регулирует слияние аутофагосома-лизосома (Jiang et al., 2014). Сходным образом UVRAG незаменим для слияния аутофагосом и лизосом у Drosophila (Takats et al., 2014). Необходимы дальнейшие исследования для уточнения функции UVRAG во время слияния.
Активный, связанный с GTP белок Rab связывается с различными эффекторами, которые обычно регулируют подвижность везикул и слияние с правильным мембранным компартментом. Недавние открытия предполагают, что PLEKHM1, который первоначально был идентифицирован как гомолог Rubicon, функционирует как эффектор Rab7 и участвует в слиянии аутофагосома-лизосома через Rab7, комплекс HOPS и LC3 и / или GABARAP (McEwan et al., 2015; Табата и др., 2010) (рис.3). Было охарактеризовано несколько других эффекторов Rab7, таких как RILP и FYCO1, оба из которых функционируют в транспорте аутофагосом (см. Выше и ниже).
Рис. 3.
Роль SNAREs в обеспечении слияния аутофагосома-лизосома. EPG5 рекрутируется в поздние эндосомы / лизосомы вместе с Rab7 и VAMP-8, где он связывает поздние эндосомы / лизосомы путем связывания с LC3 и STX17-SNAP29; это облегчает сборку комплекса trans-SNARE для слияния.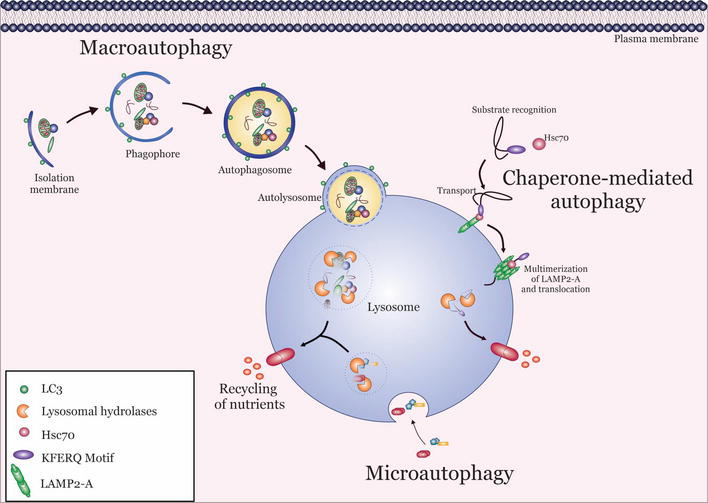 В отличие от EPG5, ATG14L связывается с промежуточными комплексами SNARE STX17 и STX17-SNAP29 Qabc на аутофагосомах, но не с комплексами STX17-SNAP29-VAMP8 RQabc SNARE, указывая тем самым, что ATG14L действует раньше, чем EPG5. PLEKHM1 является адаптерным белком, который взаимодействует с Rab7, комплексами HOPS-SNARE и белками LC3 и / или GABARAP, чтобы облегчить слияние аутофагосома-лизосома. EPG5, ATG14L и комплекс HOPS действуют как факторы привязки, но их точное соотношение все еще требует уточнения. O -GlcNAцилированный SNAP-29, который генерируется O-связанной β-N-ацетилглюкозамин (O-GlcNAc) трансферазой (OGT), имеет пониженную аффинность связывания со своими партнерскими SNARE.Эта модификация подавляется голоданием, и снижение уровней O -GlcNAцилированного SNAP-29 действует как сигнал для сборки SNAP-29-содержащих комплексов транс-SNARE, таким образом стимулируя аутофагию.
В отличие от EPG5, ATG14L связывается с промежуточными комплексами SNARE STX17 и STX17-SNAP29 Qabc на аутофагосомах, но не с комплексами STX17-SNAP29-VAMP8 RQabc SNARE, указывая тем самым, что ATG14L действует раньше, чем EPG5. PLEKHM1 является адаптерным белком, который взаимодействует с Rab7, комплексами HOPS-SNARE и белками LC3 и / или GABARAP, чтобы облегчить слияние аутофагосома-лизосома. EPG5, ATG14L и комплекс HOPS действуют как факторы привязки, но их точное соотношение все еще требует уточнения. O -GlcNAцилированный SNAP-29, который генерируется O-связанной β-N-ацетилглюкозамин (O-GlcNAc) трансферазой (OGT), имеет пониженную аффинность связывания со своими партнерскими SNARE.Эта модификация подавляется голоданием, и снижение уровней O -GlcNAцилированного SNAP-29 действует как сигнал для сборки SNAP-29-содержащих комплексов транс-SNARE, таким образом стимулируя аутофагию.
Рис. 3.
Роль SNAREs в обеспечении слияния аутофагосомы и лизосомы. EPG5 рекрутируется в поздние эндосомы / лизосомы вместе с Rab7 и VAMP-8, где он связывает поздние эндосомы / лизосомы путем связывания с LC3 и STX17-SNAP29; это облегчает сборку комплекса trans-SNARE для слияния.В отличие от EPG5, ATG14L связывается с промежуточными комплексами SNARE STX17 и STX17-SNAP29 Qabc на аутофагосомах, но не с комплексами STX17-SNAP29-VAMP8 RQabc SNARE, указывая тем самым, что ATG14L действует раньше, чем EPG5. PLEKHM1 является адаптерным белком, который взаимодействует с Rab7, комплексами HOPS-SNARE и белками LC3 и / или GABARAP, чтобы облегчить слияние аутофагосома-лизосома. EPG5, ATG14L и комплекс HOPS действуют как факторы привязки, но их точное соотношение все еще требует уточнения. O -GlcNAцилированный SNAP-29, который генерируется O-связанной β-N-ацетилглюкозамин (O-GlcNAc) трансферазой (OGT), имеет пониженную аффинность связывания со своими партнерскими SNARE.Эта модификация подавляется голоданием, и снижение уровней O -GlcNAцилированного SNAP-29 действует как сигнал для сборки SNAP-29-содержащих комплексов транс-SNARE, таким образом стимулируя аутофагию.
Известно, что в дополнение к Rab7 стадию слияния регулирует Rab33b. Rab33b представляет собой белок Rab, локализованный в комплексе Гольджи, который играет роль в образовании аутофагосом посредством взаимодействия с ATG16 (Itoh et al., 2011). Его GAP ornithine aminotransferase-like 1 (OATL1, также известный как TBC1D25) рекрутируется на аутофагосомы посредством прямого взаимодействия с ATG8, а сверхэкспрессия OATL1, как было показано, ингибирует слияние аутофагосома-лизосома (Itoh et al., 2011). Недавно было показано, что во время ремоделирования Т-канальцев у Drosophila Rab2 локализуется в завершенных аутофагосомах и взаимодействует с комплексом HOPS, чтобы способствовать слиянию аутофагосома-лизосома (Fujita et al., 2017). Эти результаты подтверждают, что, хотя Rab7 играет ключевую роль во время слияния аутофагосома-лизосома, дальнейшие малые GTPases участвуют в этом процессе и другие — пока не оцененные — функции членов этого семейства белков могут быть обнаружены в будущем.
Более 60 SNARE определяют специфичность слияния мембран и управляют процессами слияния клеток млекопитающих. Функционально SNARE сгруппированы в v-SNARE на донорских везикулах и t-SNARE на целевых везикулах. Структурно SNARE подразделяются на Q-SNARE (которые имеют аминокислотный остаток Q) и R-SNARE (которые имеют аминокислотный остаток R). Q-SNARE далее делятся на Qa-, Qb- и Qc-SNARE на основе аминокислотной последовательности домена SNARE.Эти SNARE образуют параллельный пучок с четырьмя спиралями, состоящий из Qa-, Qb-, Qc- и R-SNARE, которые соединяют две сливающиеся мембраны.
При голодании у млекопитающих синтаксин 17 Qa-SNARE (STX17) задействуется, предположительно из цитозоля в закрытые аутофагосомы, и опосредует слияние аутофагосома-лизосома путем связывания со своими партнерами, Qbc-SNARE SNAP29 и лизосомным R-SNARE V-SNARE. (Itakura et al., 2012) (рис.3). Соответственно, истощение STX17 вызывает накопление аутофагосом.Сходные механизмы и механизмы используются также для слияния аутофагосом и лизосом у Drosophila (Takats et al., 2013). Более того, O -связанная N -ацетилглюкозаминная (O-GlcNAc) модификация SNAP29 негативно регулирует SNARE-зависимое слияние аутофагосом и лизосом (Guo et al., 2014). Следовательно, нокдаун O-GlcNAc трансферазы или мутации сайтов SNAP29 O-GlcNAc способствует образованию SNAP29-содержащего комплекса SNARE и увеличивает слияние между аутофагосомами и лизосомами (рис.3).
(Itakura et al., 2012) (рис.3). Соответственно, истощение STX17 вызывает накопление аутофагосом.Сходные механизмы и механизмы используются также для слияния аутофагосом и лизосом у Drosophila (Takats et al., 2013). Более того, O -связанная N -ацетилглюкозаминная (O-GlcNAc) модификация SNAP29 негативно регулирует SNARE-зависимое слияние аутофагосом и лизосом (Guo et al., 2014). Следовательно, нокдаун O-GlcNAc трансферазы или мутации сайтов SNAP29 O-GlcNAc способствует образованию SNAP29-содержащего комплекса SNARE и увеличивает слияние между аутофагосомами и лизосомами (рис.3).
Недавно было показано, что изоляционная мембрана формируется в месте контакта ER-митохондрий и STX17 также участвует в этом процессе (Hamasaki et al., 2013). STX17 локализуется в ER в условиях питания, но после голодания он перемещается в сайты контакта ER-митохондрий (Hamasaki et al., 2013). Здесь STX17 связывается и рекрутирует ATG14L в контакты ER-митохондрий, чтобы инициировать образование изолирующей мембраны.Напротив, STX17, который участвует в слиянии аутофагосом и лизосом, предположительно рекрутируется из цитозоля в закрытые аутофагосомы (Itakura et al., 2012). Необходимы дальнейшие исследования, чтобы понять, как клетки используют разные пулы STX17 в зависимости от контекста (ER или цитоплазма). Помимо STX17, ATG14L связывается с бинарным комплексом, образованным между STX17 и SNAP29 (STX17-SNAP29), и способствует его взаимодействию с VAMP8, способствуя слиянию аутофагосома-лизосома (Diao et al., 2015) (рис.3), что указывает на взаимодействие между STX17 и ATG14L как на ранних, так и на поздних стадиях аутофагии.
Мембранные привязи, как полагают, обеспечивают другой уровень специфичности и облегчают стыковку и слияние, перекрывая противоположные мембраны и стимулируя образование комплекса SNARE. В дополнение к своей хорошо известной роли в пути эндоцитоза, комплекс HOPS действует как фактор привязки для слияния аутофагосом (Jiang et al., 2014) (рис.3). STX17 взаимодействует с комплексом HOPS, включая VPS33A, VPS16, VPS11, VPS18, VPS39 и VPS41. В соответствии с этим, эти субъединицы HOPS рекрутируются в STX17-положительные аутофагосомы при голодании. Важно отметить, что нокдаун VPS33A, VPS16 или VPS39 блокирует аутофагический поток и вызывает накопление STX17- и LC3-позитивных аутофагосом, подтверждая, что HOPS способствует слиянию аутофагосома-лизосома с STX17 (Jiang et al., 2014). Большинство этих находок также наблюдается у Drosophila (Takats et al., 2014). Недавнее структурное исследование Vps33 у термофильных грибов Chaetomium thermophilum подтвердило, что Vps33 способствует сборке SNARE за счет точного позиционирования и выравнивания SNAREs (Baker et al., 2015). Это открытие дает нам новое понимание того, как комплекс HOPS способствует сборке SNARE.
В дополнение к своей хорошо известной роли в пути эндоцитоза, комплекс HOPS действует как фактор привязки для слияния аутофагосом (Jiang et al., 2014) (рис.3). STX17 взаимодействует с комплексом HOPS, включая VPS33A, VPS16, VPS11, VPS18, VPS39 и VPS41. В соответствии с этим, эти субъединицы HOPS рекрутируются в STX17-положительные аутофагосомы при голодании. Важно отметить, что нокдаун VPS33A, VPS16 или VPS39 блокирует аутофагический поток и вызывает накопление STX17- и LC3-позитивных аутофагосом, подтверждая, что HOPS способствует слиянию аутофагосома-лизосома с STX17 (Jiang et al., 2014). Большинство этих находок также наблюдается у Drosophila (Takats et al., 2014). Недавнее структурное исследование Vps33 у термофильных грибов Chaetomium thermophilum подтвердило, что Vps33 способствует сборке SNARE за счет точного позиционирования и выравнивания SNAREs (Baker et al., 2015). Это открытие дает нам новое понимание того, как комплекс HOPS способствует сборке SNARE.
Как комплексы HOPS рекрутируются в аутофагосомы и лизосомы? На поздних эндосомах и лизосомах Rab7 рекрутирует свои эффекторы PLEKHM1 и RILP, которые связываются с компонентами HOPS VPS39 и VPS41 соответственно (Wijdeven et al., 2016). Эти эффекторы, таким образом, совместно привлекают комплекс HOPS для слияния. Это похоже на дрожжи, где Rab7 гомолог Ypt7 приобретает HOPS путем связывания как с Vps39, так и с Vps41 (Ostrowicz et al., 2010; Plemel et al., 2011). Помимо PLEKHM1 и RILP, Rab7 задействует другой эффектор, датчик холестерина ORP1L, который связывается с Rab7 в присутствии RILP и негативно регулирует слияние (Johansson et al., 2007; Wijdeven et al., 2016). При низких уровнях холестерина ORP1L на поздних эндосомах или лизосомах взаимодействует с ER-белком VAP-A с образованием сайта контакта ER-аутофагосомы.Этот контактный сайт предотвращает рекрутирование PLEKHM1 на Rab7 и, следовательно, комплекса HOPS, что приводит к дефекту слияния аутофагосома-поздняя эндосома / лизосома. Интересно, что ORP1L и присутствие этого контактного сайта предотвращают рекрутирование dynein и / или dynactin с помощью RILP и, таким образом, направленный на минус-конец транспорт поздних аутофагосом (Wijdeven et al., 2016). Эти наблюдения предполагают, что слияние аутофагосом с поздними эндосомами и лизосомами и транспорт поздних аутофагосом регулируются холестерином.Однако функция in vivo холестерина во время слияния все еще неясна и требует рассмотрения в будущих исследованиях.
Интересно, что ORP1L и присутствие этого контактного сайта предотвращают рекрутирование dynein и / или dynactin с помощью RILP и, таким образом, направленный на минус-конец транспорт поздних аутофагосом (Wijdeven et al., 2016). Эти наблюдения предполагают, что слияние аутофагосом с поздними эндосомами и лизосомами и транспорт поздних аутофагосом регулируются холестерином.Однако функция in vivo холестерина во время слияния все еще неясна и требует рассмотрения в будущих исследованиях.
Ectopic P granules белок 5 (EPG5) был первоначально идентифицирован в ходе генетического скрининга C. elegans и является еще одним эффектором Rab7 и связывающим фактором, который определяет специфичность слияния аутофагосом с эндосомами / лизосомами (Tian et al., 2010; Wang et al., др., 2016) (рис.3). EPG5 рекрутируется в поздние эндосомы / лизосомы посредством прямого взаимодействия с Rab7 и поздним эндосомным / лизосомным R-SNARE VAMP7 / VAMP8. EPG5 также связывается с LC3 / LGG-1 (LGG-1 является гомологом C. elegans Atg8) через мотив LC3-взаимодействующей области (LIR) и с собранными комплексами STX17-SNAP29 (Qabc-SNARE) на аутофагосомах. EPG5 стабилизирует и облегчает сборку транс-SNARE комплексов STX17-SNAP29-VAMP8 и, таким образом, наиболее вероятно способствует слиянию между аутофагосомами и лизосомами.Потеря EPG5 вызывает аномальное слияние аутофагосом с различными эндоцитарными пузырьками, частично из-за повышенной сборки комплекса STX17-SNAP25-VAMP8. В соответствии с этим, нокдаун SNAP25 частично подавляет влияние на аутофагию из-за истощения EPG5 (Wang et al., 2016). Эти находки, таким образом, начали отвечать на вопрос, почему аутофагосомы специфически сливаются с поздними эндосомами и лизосомами. В дополнение к HOPS и EPG5, как упоминалось ранее, ATG14L непосредственно связывается с бинарным комплексом STX17-SNAP29 на аутофагосомах и способствует слиянию аутофагосом, опосредованному STX17-SNAP29-VAMP8, с лизосомами, таким образом функционируя как фактор привязки (Diao et al. , 2015) (рис.3). Однако точная взаимосвязь между HOPS, EPG5 и ATG14L все еще неясна и требует уточнения в будущих исследованиях. Tectonin beta-propeller repeat-containing 1 (TECPR1) также функционирует как фактор привязки, и мы обсудим его роль далее в тексте (Chen et al., 2012).
, 2015) (рис.3). Однако точная взаимосвязь между HOPS, EPG5 и ATG14L все еще неясна и требует уточнения в будущих исследованиях. Tectonin beta-propeller repeat-containing 1 (TECPR1) также функционирует как фактор привязки, и мы обсудим его роль далее в тексте (Chen et al., 2012).
Фосфоинозитиды (ИП) участвуют в перемещении внутриклеточной мембраны. Фосфорилирование третьего, четвертого и пятого положения инозитолового кольца PI дает различные варианты, в частности PI3P [фосфатидилинозитол (3) -фосфат], который играет хорошо изученную роль в образовании аутофагосом.Хотя большинство исследований сосредоточено на белках, которые участвуют в процессе аутофагии, несколько недавних исследований выявили роль ИП на поздней стадии аутофагии, включая транспорт аутофагосом и слияние аутофагосом с лизосомами.
Исследования дрожжей дают подсказки о том, как аутофагосомы становятся способными сливаться с лизосомами. Дефосфорилирование и клиренс PI3P фосфатазой PI3P Ymr1 на завершенной аутофагосоме важны для слияния аутофагосомы и вакуоли (Cebollero et al., 2012). Очистка PI3P запускает диссоциацию аппарата ATG от внешней мембраны аутофагосомы, что делает аутофагосому компетентной для слияния. Напротив, формы фосфатаз PI3P млекопитающих, такие как миотубулярный белок 3 (MTMR3) и MTMR14 (также известный как Jumpy), по-видимому, выполняют разные функции во время аутофагии (Taguchi-Atarashi et al., 2010; Vergne et al., 2009 г.). Нокдаун MTMR3 и MTMR14 увеличивает образование аутофагосом, указывая на то, что эти фосфатазы являются негативными регуляторами аутофагии и что они действуют на ранних стадиях образования аутофагосом.Таким образом, стоит дополнительно исследовать, выполняют ли другие PI фосфатазы аналогичные функции у млекопитающих.
Как упоминалось выше, FYCO1 связывается с LC3, PI3P и Rab7 и участвует в движении аутофагосом (Рис. 2). Эндогенный FYCO1 локализуется на перинуклеарных цитозольных пузырьках, но после голодания он также распределяется по периферии клеток микротрубочко-зависимым образом (Pankiv et al., 2010).FYCO1 функционирует как адаптерный белок между аутофагосомами и молекулярными моторами, направленными на плюс-концы микротрубочек — о чем свидетельствуют истощенные FYCO1 клетки, которые накапливают аутофагосомы в перинуклеарных кластерах (Pankiv et al., 2010). Недавно было показано, что FYCO1 на эндосомах взаимодействует непосредственно с легкой цепью KLC2 kinesin1 во время транслокации эндосом на периферию клеток (Raiborg et al., 2015). Таким образом, представляется целесообразным изучить, используются ли одни и те же кинезины во время транспорта аутофагосом.
2). Эндогенный FYCO1 локализуется на перинуклеарных цитозольных пузырьках, но после голодания он также распределяется по периферии клеток микротрубочко-зависимым образом (Pankiv et al., 2010).FYCO1 функционирует как адаптерный белок между аутофагосомами и молекулярными моторами, направленными на плюс-концы микротрубочек — о чем свидетельствуют истощенные FYCO1 клетки, которые накапливают аутофагосомы в перинуклеарных кластерах (Pankiv et al., 2010). Недавно было показано, что FYCO1 на эндосомах взаимодействует непосредственно с легкой цепью KLC2 kinesin1 во время транслокации эндосом на периферию клеток (Raiborg et al., 2015). Таким образом, представляется целесообразным изучить, используются ли одни и те же кинезины во время транспорта аутофагосом.
В дополнение к комплексу HOPS, PI3P-связывающий белок TECPR1, как полагают, действует как фактор привязки при слиянии аутофагосом (Chen et al., 2012) (Fig. 4). TECPR1-истощенные клетки нарушают аутофагический поток и накапливают аутофагические вакуоли и субстраты, включая p62 (официально известный как SQSTM1) и липидированный LC3 (LC3-II) (Chen et al., 2012). Первоначально TECPR1 был идентифицирован благодаря его взаимодействию с ATG5 (Behrends et al., 2010). В то время как локализация комплекса Atg12-Atg5-Atg16 в фагофоре (изолирующей мембране) зависит от присутствия PI3P, Chen et al. показывают, что TECPR1 и ATG16 образуют взаимоисключающие комплексы с конъюгатом ATG12-ATG5, а TECPR1 связывает PI3P при ассоциации с конъюгатом Atg12-Atg5 (Chen et al., 2012; Fujita et al., 2008b). Более того, TECPR1 локализуется в лизосомах / аутолизосомах и рекрутирует конъюгат ATG12-ATG5, который делает возможным его связывание с PI3P, тем самым, возможно, способствуя созреванию аутофагосом и слиянию аутофагосом и лизосом.Однако другое исследование обнаружило, что TECPR1 также участвует в биогенезе и созревании фагофоров во время избирательной аутофагии (Ogawa et al.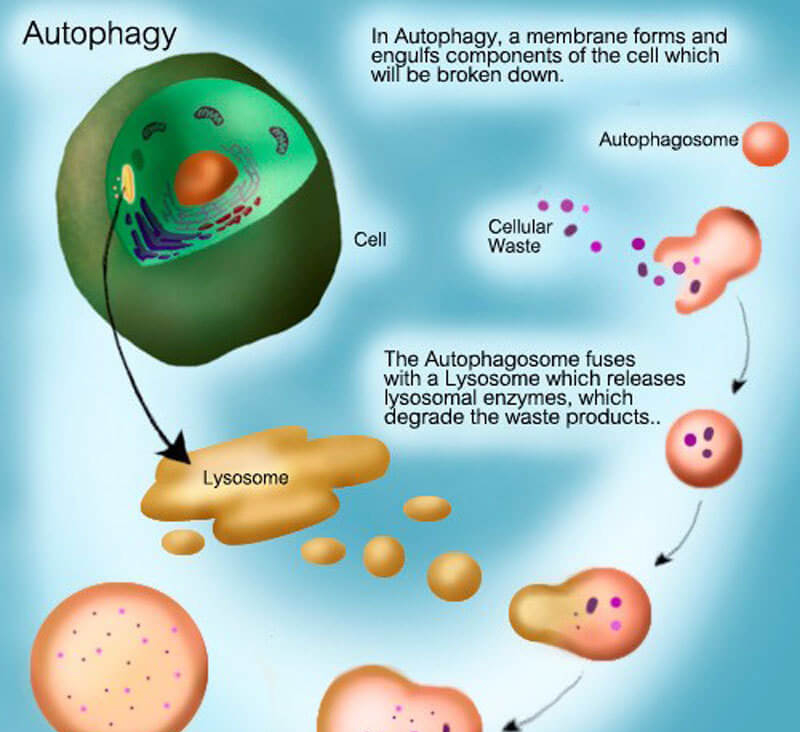 , 2011). Эти несоответствия могут отражать либо двойную роль TECPR1, либо различные функции в разных биологических контекстах. Более того, функциональная взаимосвязь между несколькими вышеупомянутыми факторами привязки все еще неясна и требует решения.
, 2011). Эти несоответствия могут отражать либо двойную роль TECPR1, либо различные функции в разных биологических контекстах. Более того, функциональная взаимосвязь между несколькими вышеупомянутыми факторами привязки все еще неясна и требует решения.
Рис. 4.
Роль фосфолипидов в процессе слияния аутофагосома – лизосома. Определенное количество клеточного INPP5E локализуется в лизосомах, где он превращает PI (3,5) P2 в PI3P, что затем приводит к фосфорилированию и активации корактина (CTTN). Активированный CTTN важен для стабилизации актиновых филаментов и, следовательно, для слияния аутофагосома-лизосома. TECPR1 на лизосомах функционирует как связывающий фактор, который взаимодействует с ATG12 – ATG5 и PI3P на аутофагосомах. GABARAP рекрутирует пальмитоилированный PI4KIIα из сети транс-Гольджи в аутофагосомы, PI4P, который продуцируется с помощью PI4KIIα, необходим для слияния, хотя его точная функция все еще неизвестна.
Рис. 4.
Роль фосфолипидов в процессе слияния аутофагосомы и лизосомы. Определенное количество клеточного INPP5E локализуется в лизосомах, где он превращает PI (3,5) P2 в PI3P, что затем приводит к фосфорилированию и активации корактина (CTTN). Активированный CTTN важен для стабилизации актиновых филаментов и, следовательно, для слияния аутофагосома-лизосома. TECPR1 на лизосомах функционирует как связывающий фактор, который взаимодействует с ATG12 – ATG5 и PI3P на аутофагосомах.GABARAP рекрутирует пальмитоилированный PI4KIIα из сети транс-Гольджи в аутофагосомы, PI4P, который продуцируется с помощью PI4KIIα, необходим для слияния, хотя его точная функция все еще неизвестна.
Генерация PI4P [phosphatidylinositol (4) -phosphate] на аутофагосомах является критической для слияния аутофагосома-лизосома у млекопитающих (Wang et al. , 2015) (Fig. 4). Фосфатидилинозитол-4-киназа типа 2-альфа (PI4KIIα) обычно локализуется в перинуклеарной области и транс-сети Гольджи (TGN).После голодания PI4KIIα выходит из TGN и диспергируется в цитоплазме, при этом часть PI4KIIα локализуется в аутофагосомах зависимым от пальмитоилирования способом с аналогичным перераспределением PI4P (Wang et al., 2015). В соответствии с этим истощение PI4KIIα снижает концентрацию PI4P в аутофагосомах. Интересно, что PI4KIIα взаимодействует с гомологами Atg8 GABARAP и GABARAPL2, но не с LC3. Истощение GABARAP ингибирует транслокацию PI4KIIα в аутофагосомы, тогда как истощение PI4KIIα не влияет на распределение GABARAP, что указывает на то, что GABARAP функционирует выше PI4KIIα в этом контексте (Wang et al., 2015). Нокдаун либо GABARAP, либо PI4KIIα создает дефектные большие аутофагосомы и нарушает деградацию LC3 и p62 из-за дефектного слияния аутофагосом с лизосомами. Важно отметить, что этот дефект слияния устраняется введением PI4P, но не фосфатидилинозитол (4,5) -бисфосфата, что позволяет предположить, что генерация именно PI4P — а не его последующих метаболитов — важна для слияния аутофагосома-лизосома (Wang et al. , 2015). Однако точная роль PI4P в процессе слияния нуждается в уточнении в будущих исследованиях.В соответствии с функцией GABARAP во время слияния аутофагосома-лизосома, недавний систематический функциональный анализ семейства Atg8 у млекопитающих показал, что подсемейство GABARAP способствует привлечению PLEKHM1 и управляет слиянием аутофагосома-лизосома, тогда как подсемейство LC3 играет менее важную роль в именно эти процессы (Nguyen et al., 2016).
, 2015) (Fig. 4). Фосфатидилинозитол-4-киназа типа 2-альфа (PI4KIIα) обычно локализуется в перинуклеарной области и транс-сети Гольджи (TGN).После голодания PI4KIIα выходит из TGN и диспергируется в цитоплазме, при этом часть PI4KIIα локализуется в аутофагосомах зависимым от пальмитоилирования способом с аналогичным перераспределением PI4P (Wang et al., 2015). В соответствии с этим истощение PI4KIIα снижает концентрацию PI4P в аутофагосомах. Интересно, что PI4KIIα взаимодействует с гомологами Atg8 GABARAP и GABARAPL2, но не с LC3. Истощение GABARAP ингибирует транслокацию PI4KIIα в аутофагосомы, тогда как истощение PI4KIIα не влияет на распределение GABARAP, что указывает на то, что GABARAP функционирует выше PI4KIIα в этом контексте (Wang et al., 2015). Нокдаун либо GABARAP, либо PI4KIIα создает дефектные большие аутофагосомы и нарушает деградацию LC3 и p62 из-за дефектного слияния аутофагосом с лизосомами. Важно отметить, что этот дефект слияния устраняется введением PI4P, но не фосфатидилинозитол (4,5) -бисфосфата, что позволяет предположить, что генерация именно PI4P — а не его последующих метаболитов — важна для слияния аутофагосома-лизосома (Wang et al. , 2015). Однако точная роль PI4P в процессе слияния нуждается в уточнении в будущих исследованиях.В соответствии с функцией GABARAP во время слияния аутофагосома-лизосома, недавний систематический функциональный анализ семейства Atg8 у млекопитающих показал, что подсемейство GABARAP способствует привлечению PLEKHM1 и управляет слиянием аутофагосома-лизосома, тогда как подсемейство LC3 играет менее важную роль в именно эти процессы (Nguyen et al., 2016).
В отличие от очевидной роли фосфолипидов в мембранах аутофагосом, было неясно, важен ли конкретный состав фосфолипидов в лизосомной мембране для слияния аутофагосом и лизосом.Но недавно мы идентифицировали фосфоинозитид-фосфатазу инозитолполифосфат-5-фосфатазу E (INPP5E) как до сих пор неизвестный регулятор аутофагии, который способствует стадии слияния (Hasegawa et al.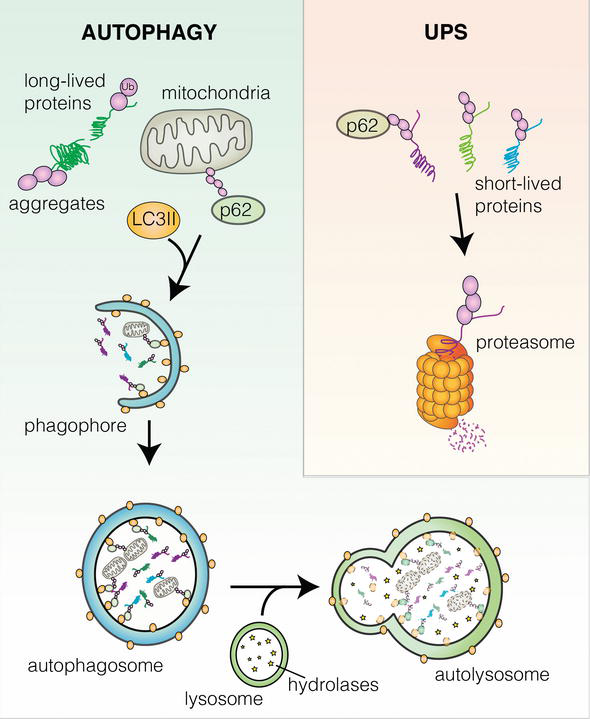 , 2016; Nakamura et al., 2016) (рис. . 4). Ингибирование INPP5E вызывает накопление аутофагосом, как определено с помощью LC3 puncta, путем нарушения слияния аутофагосома-лизосома. Важно отметить, что нокдаун INPP5E не влияет на эндоцитарный путь или целостность лизосом, дополнительно подчеркивая специфическую роль INPP5E во время стадии слияния аутофагосома-лизосома.Примечательно, что часть белка INPP5E локализуется на мембранах лизосом, где он опосредует превращение фосфатидилинозитол (3,5) -бисфосфата [PI (3,5) P2] в PI3P, что имеет решающее значение для слияния с аутофагосомами. Нокдаун 1-фосфатидилинозитол-3-фосфат-5-киназы (PIKFYVE) снижает уровни PI (3,5) P2 в лизосомах и вызывает серьезные дефекты аутофагии из-за потери лизосомной функции (de Lartigue et al., 2009). Следовательно, правильные уровни PI (3,5) P2 в лизосомной мембране, по-видимому, необходимы для функции лизосом и для их слияния с аутофагосомами во время аутофагии.Активность INPP5E в отношении лизосом, как было показано, в конечном итоге необходима для фосфорилирования cortactin (CTTN), что ведет к полимеризации актина с последующим слиянием аутофагосома-лизосома (Hasegawa et al., 2016) (Fig. 4). Мутации в INPP5E связаны с синдромом Жубера, редким заболеванием мозга (Bielas et al., 2009). Интересно, что эти мутантные формы INPP5E не устраняют дефекты аутофагии, которые вызваны нокдауном INPP5E в нейрональных клетках, что позволяет предположить, что нарушение аутофагии вызывает это заболевание (Hasegawa et al., 2016). Поскольку INPP5E преимущественно экспрессируется в нейрональных клетках, необходимы будущие исследования, чтобы выяснить, участвует ли подобный механизм в слиянии аутофагосома-лизосома в др. Типах клеток.
, 2016; Nakamura et al., 2016) (рис. . 4). Ингибирование INPP5E вызывает накопление аутофагосом, как определено с помощью LC3 puncta, путем нарушения слияния аутофагосома-лизосома. Важно отметить, что нокдаун INPP5E не влияет на эндоцитарный путь или целостность лизосом, дополнительно подчеркивая специфическую роль INPP5E во время стадии слияния аутофагосома-лизосома.Примечательно, что часть белка INPP5E локализуется на мембранах лизосом, где он опосредует превращение фосфатидилинозитол (3,5) -бисфосфата [PI (3,5) P2] в PI3P, что имеет решающее значение для слияния с аутофагосомами. Нокдаун 1-фосфатидилинозитол-3-фосфат-5-киназы (PIKFYVE) снижает уровни PI (3,5) P2 в лизосомах и вызывает серьезные дефекты аутофагии из-за потери лизосомной функции (de Lartigue et al., 2009). Следовательно, правильные уровни PI (3,5) P2 в лизосомной мембране, по-видимому, необходимы для функции лизосом и для их слияния с аутофагосомами во время аутофагии.Активность INPP5E в отношении лизосом, как было показано, в конечном итоге необходима для фосфорилирования cortactin (CTTN), что ведет к полимеризации актина с последующим слиянием аутофагосома-лизосома (Hasegawa et al., 2016) (Fig. 4). Мутации в INPP5E связаны с синдромом Жубера, редким заболеванием мозга (Bielas et al., 2009). Интересно, что эти мутантные формы INPP5E не устраняют дефекты аутофагии, которые вызваны нокдауном INPP5E в нейрональных клетках, что позволяет предположить, что нарушение аутофагии вызывает это заболевание (Hasegawa et al., 2016). Поскольку INPP5E преимущественно экспрессируется в нейрональных клетках, необходимы будущие исследования, чтобы выяснить, участвует ли подобный механизм в слиянии аутофагосома-лизосома в др. Типах клеток.
Хотя несколько PIs, как в аутофагосоме, так и в лизосоме, участвуют в слиянии аутофагосома-лизосома, будущие исследования должны изучить точную роль каждого PI, временную шкалу функции PI и то, как разные PIs функционируют совместно во время процесса слияния.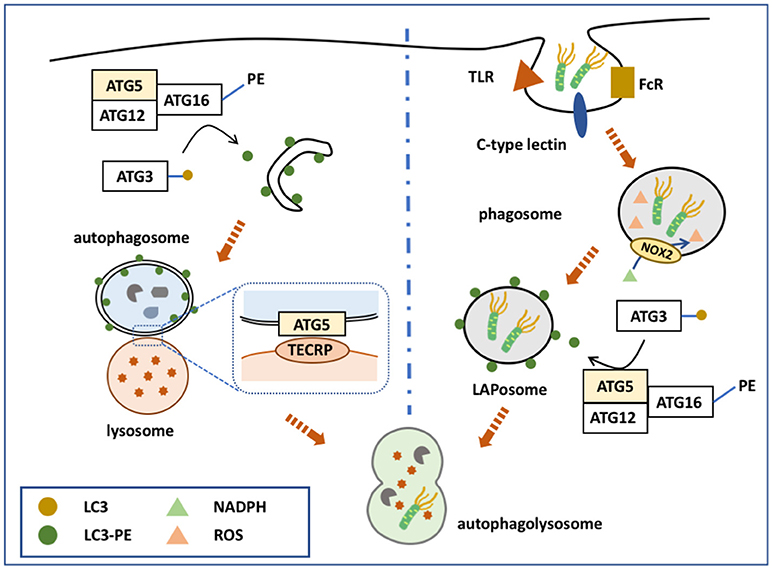
Формирование аутофагосом по отношению к эндоплазматическому ретикулуму | Журнал биомедицинских наук
Дикич И., Элазар З. Механизм и медицинские последствия аутофагии млекопитающих. Nat Rev Mol Cell Biol. 2018; 19 (6): 349–64.
CAS PubMed Статья PubMed Central Google Scholar
Мидзусима Н. Аутофагия: процесс и функция. Genes Dev. 2007. 21 (22): 2861–73.
CAS PubMed Статья PubMed Central Google Scholar
Мелия Т.Дж., Листад А.Х., Симонсен А. Биогенез аутофагосом: от роста мембраны до закрытия. J Cell Biol. 2020; 219 (6): e202002085.
PubMed Статья CAS PubMed Central Google Scholar
Мидзусима Н., Йошимори Т., Осуми Ю. Роль белков Atg в формировании аутофагосом.Annu Rev Cell Dev Biol. 2011. 27 (1): 107–32.
CAS PubMed Статья PubMed Central Google Scholar
Накатогава Х. Механизмы, регулирующие биогенез аутофагосом. Nat Rev Mol Cell Biol. 2020; 21 (8): 439–58.
CAS PubMed Статья PubMed Central Google Scholar
Кобаяси С., Кодзидани Т., Осакада Х., Ямамото А., Йошимори Т., Хираока Ю. и др.Искусственная индукция аутофагии вокруг шариков полистирола в нефагоцитарных клетках. Аутофагия. 2010. 6 (1): 36–45.
CAS PubMed Статья PubMed Central Google Scholar
Mi N, Chen Y, Wang S, Chen M, Zhao M, Yang G и др. CapZ регулирует формирование аутофагосомной мембраны, способствуя сборке актина внутри изолирующей мембраны. Nat Cell Biol. 2015; 17 (9): 1112–23.
CAS PubMed Статья PubMed Central Google Scholar
Сингх С.Б., Дэвис А.С., Тейлор Г.А., Деретик В. Человеческий IRGM вызывает аутофагию для устранения внутриклеточных микобактерий. Наука. 2006. 313 (5792): 1438–41.
CAS PubMed Статья PubMed Central Google Scholar
Ямагути Х., Накагава И., Ямамото А., Амано А., Нода Т., Йошимори Т. Начальная стадия образования ГАЗ-содержащих аутофагосом подобных вакуолей требует Rab7. PLoS Pathog. 2009; 5 (11): e1000670.
PubMed PubMed Central Статья CAS Google Scholar
Джин М., Клионский DJ. Регулирование аутофагии: Модуляция размера и количества аутофагосом. FEBS Lett. 2014. 588 (15): 2457–63.
CAS PubMed PubMed Central Статья Google Scholar
Xie Z, Nair U, Klionsky DJ. Atg8 контролирует экспансию фагофоров во время образования аутофагосом. MBoC. 2008. 19 (8): 3290–8.
CAS PubMed Статья Google Scholar
Ямамото Ю., Касаи А., Омори Х., Такино Т., Сугихара М., Умемото Т. и др. ERdj8 определяет размер аутофагосом в процессе формирования. J Cell Biol. 2020; 219 (8): e201
7.CAS PubMed Статья Google Scholar
Анелли Т., Сития Р. Контроль качества белка на раннем секреторном пути. EMBO J. 2008; 27 (2): 315–27.
CAS PubMed PubMed Central Статья Google Scholar
Johnson AE, van Waes MA. Транслокон: динамический шлюз на мембране ER. Annu Rev Cell Dev Biol. 1999. 15 (1): 799–842.
PubMed Статья PubMed Central Google Scholar
Вембар СС, Бродский Ж.Л. Шаг за шагом: деградация, связанная с эндоплазматическим ретикулумом. Nat Rev Mol Cell Biol. 2008. 9 (12): 944–57.
2008. 9 (12): 944–57.
CAS PubMed PubMed Central Статья Google Scholar
Лев С. Невезикулярный перенос липидов из эндоплазматического ретикулума. Cold Spring Harb Perspect Biol. 2012; 4 (10): a013300.
PubMed PubMed Central Статья CAS Google Scholar
Берридж MJ, Bootman MD, Roderick HL. Передача сигналов кальция: динамика, гомеостаз и ремоделирование. Nat Rev Mol Cell Biol. 2003. 4 (7): 517–29.
CAS PubMed Статья PubMed Central Google Scholar
Nixon-Abell J, Obara CJ, Weigel AV, Li D, Legant WR, Xu CS и др. Повышенное пространственно-временное разрешение выявляет высокодинамичные плотные трубчатые матрицы в периферической ER. Наука. 2016; 28: 354.
Google Scholar
Воельц Г.К., Принц В.А., Шибата Ю., Рист Дж.М., Рапопорт Т.А. Класс мембранных белков, формирующих эндоплазматический ретикулум канальцев. Клетка. 2006. 124 (3): 573–86.
CAS PubMed Статья PubMed Central Google Scholar
Ван С, Тукачинский Н, Романо ФБ, Рапопорт Т.А. Взаимодействие ER-формирующих белков атластина, лунапарка и ретикулонов с образованием трубчатой мембранной сети. eLife. 2016; 5: 18605.
Артикул Google Scholar
Шибата Ю., Шемеш Т., Принц В.А., Палаццо А.Ф., Козлов М.М., Рапопорт Т.А. Механизмы, определяющие морфологию периферического ЭПР. Клетка. 2010. 143 (5): 774–88.
CAS PubMed PubMed Central Статья Google Scholar
Чанг Дж., Ли С., Блэкстоун С. Протрудин связывает атластины и белки, формирующие эндоплазматический ретикулум, и регулирует формирование сети. Proc Natl Acad Sci USA. 2013. 110 (37): 14954–9.
Proc Natl Acad Sci USA. 2013. 110 (37): 14954–9.
CAS PubMed Статья Google Scholar
Орсо Дж., Пендин Д., Лю С., Тозетто Дж., Мосс Т. Дж., Фауст Дж. Э. и др. Гомотипическое слияние мембран ER требует динамин-подобного GTPase Atlastin. Природа. 2009. 460 (7258): 978–83.
CAS PubMed Статья Google Scholar
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Формирование аутофагосом из мембранных компартментов, обогащенных фосфатидилинозитол-3-фосфатом и динамически связанных с эндоплазматическим ретикулумом. J Cell Biol. 2008. 182 (4): 685–701.
CAS PubMed PubMed Central Статья Google Scholar
Мацунага К., Морита Е., Сайто Т., Акира С., Ктистакис Н.Т., Изуми Т. и др. Аутофагия требует нацеливания на эндоплазматический ретикулум комплекса PI3-киназы через Atg14L.J Cell Biol. 2010. 190 (4): 511–21.
CAS PubMed PubMed Central Статья Google Scholar
Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T. и др. Модуляция локальных уровней PtdIns3P с помощью фосфатазы PI MTMR3 регулирует конститутивную аутофагию. Движение. 2010. 11 (4): 468–78.
CAS PubMed Статья Google Scholar
Хаяси-Нишино М., Фудзита Н., Нода Т., Ямагути А., Йошимори Т., Ямамото А. Субдомен эндоплазматического ретикулума образует колыбель для образования аутофагосом. Nat Cell Biol. 2009. 11 (12): 1433–7.
CAS PubMed Статья Google Scholar
Уэмура Т., Ямамото М., Каметака А., Су Y, Ябаши А., Ямада А. и др. Группа тонких трубчатых структур опосредует трансформацию эндоплазматического ретикулума в аутофагическую изоляционную мембрану. Mol Cell Biol. 2014. 34 (9): 1695–706.
Mol Cell Biol. 2014. 34 (9): 1695–706.
PubMed PubMed Central Статья CAS Google Scholar
Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L. Трехмерная томография выявляет связи между фагофором и эндоплазматическим ретикулумом. Аутофагия. 2009. 5 (8): 1180–5.
PubMed Статья Google Scholar
Graef M, Friedman JR, Graham C, Babu M, Nunnari J.Сайты выхода ER являются физическими и функциональными основными компонентами биогенеза аутофагосомы. MBoC. 2013. 24 (18): 2918–31.
CAS PubMed Статья PubMed Central Google Scholar
Suzuki K, Akioka M, Kondo-Kakuta C, Yamamoto H, Ohsumi Y. Тонкое картирование белков, связанных с аутофагией, во время образования аутофагосом в Saccharomyces cerevisiae . J Cell Sci. 2013; 126 (11): 2534–44.
CAS PubMed Статья PubMed Central Google Scholar
Баба М., Томонага С., Сузуки М., Генерал М., Такеда Е., Мацуура А. и др. Структура, происходящая из ядерной мембраны, ассоциированная с Atg8, участвует в секвестрации селективного груза, комплекса Cvt, во время образования аутофагосом у дрожжей. Аутофагия. 2019; 15 (3): 423–37.
CAS PubMed Статья PubMed Central Google Scholar
Каранасиос Э., Уокер С.А., Оккенхауг Х., Манифава М, Хаммель Э., Циммерманн Х. и др.Инициирование аутофагии за счет сборки комплекса ULK на тубуловезикулярных областях ER, отмеченных пузырьками ATG9. Nat Commun. 2016; 11 (7): 12420.
Артикул CAS Google Scholar
Котани Т., Кирисако Х., Коидзуми М., Осуми Ю., Накатогава Х. Комплекс Atg2-Atg18 связывает преаутофагосомные мембраны с эндоплазматическим ретикулумом для образования аутофагосом.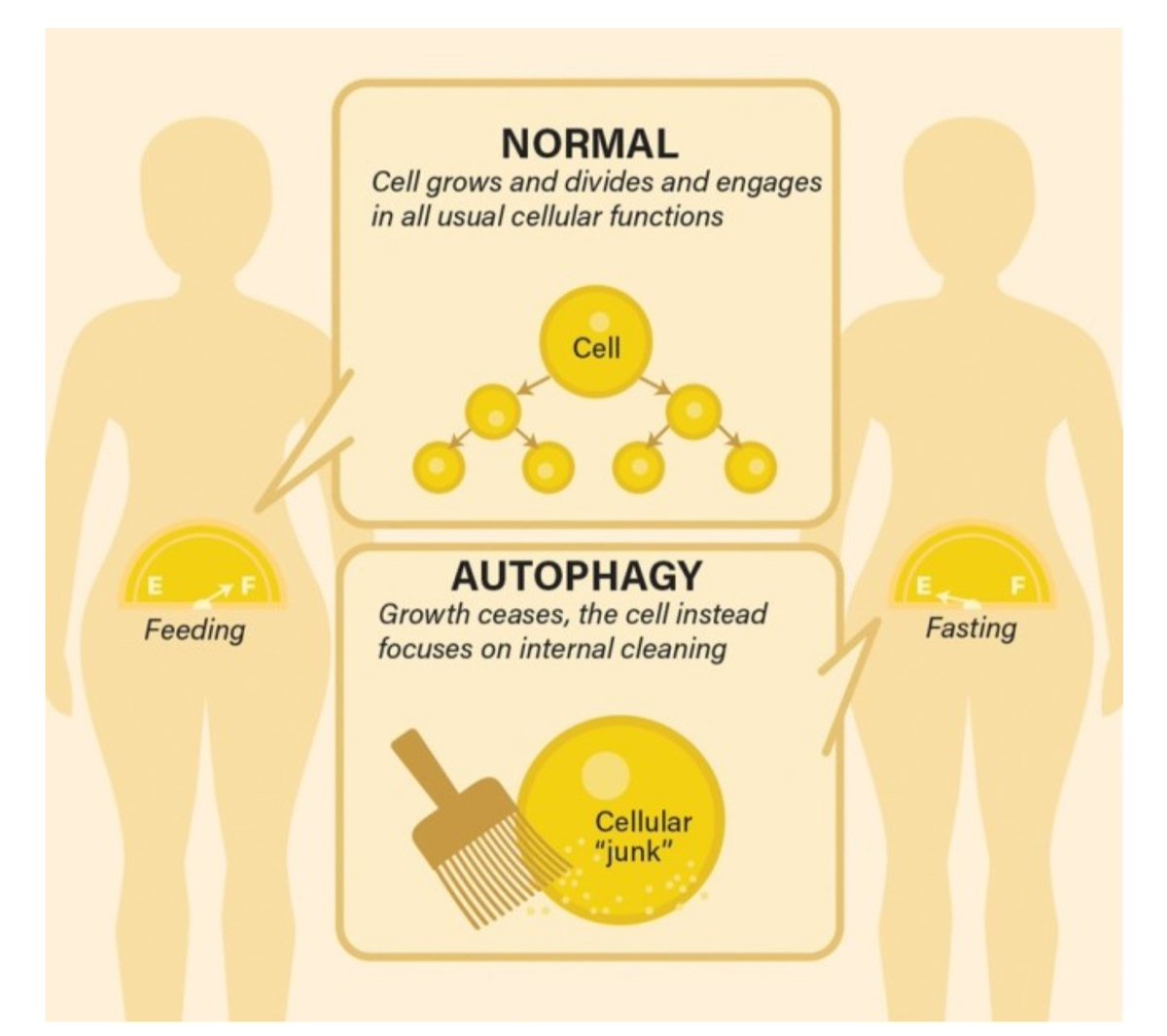 PNAS. 2018; 115 (41): 10363–8.
PNAS. 2018; 115 (41): 10363–8.
CAS PubMed Статья PubMed Central Google Scholar
Вальверде Д.П., Ю С., Боггаварапу В., Кумар Н., Лис Дж. А., Вальц Т. и др. ATG2 транспортирует липиды, способствуя биогенезу аутофагосом. J Cell Biol. 2019; 218 (6): 1787–98.
CAS PubMed PubMed Central Статья Google Scholar
Гомес-Санчес Р., Роуз Дж., Гимарайнш Р., Мари М., Папински Д., Ритер Э. и др. Atg9 устанавливает Atg2-зависимые участки контакта между эндоплазматическим ретикулумом и фагофорами. J Cell Biol.2018; 217 (8): 2743–63.
PubMed PubMed Central Статья CAS Google Scholar
Кумар Н., Леонцино М., Хэнкок-Серутти В., Хоренкамп Ф.А., Ли П., Лис Дж. А. и др. VPS13A и VPS13C представляют собой липидные транспортные белки, по-разному локализованные в сайтах контакта с ER. J Cell Biol. 2018; 217 (10): 3625–39.
CAS PubMed PubMed Central Статья Google Scholar
Маеда С., Отомо С., Отомо Т. Аутофагическая мембранная связка ATG2A переносит липиды между мембранами. eLife. 2019; 8: e45777.
PubMed PubMed Central Статья Google Scholar
Осава Т., Котани Т., Каваока Т., Хирата Е., Сузуки К., Накатогава Н. и др. Atg2 обеспечивает прямой перенос липидов между мембранами для образования аутофагосом. Nat Struct Mol Biol. 2019; 26 (4): 281–8.
CAS PubMed Статья PubMed Central Google Scholar
English AR, Voeltz GK. Rab10 GTPase регулирует динамику и морфологию ER. Nat Cell Biol. 2013; 15 (2): 169–78.
CAS PubMed Статья PubMed Central Google Scholar
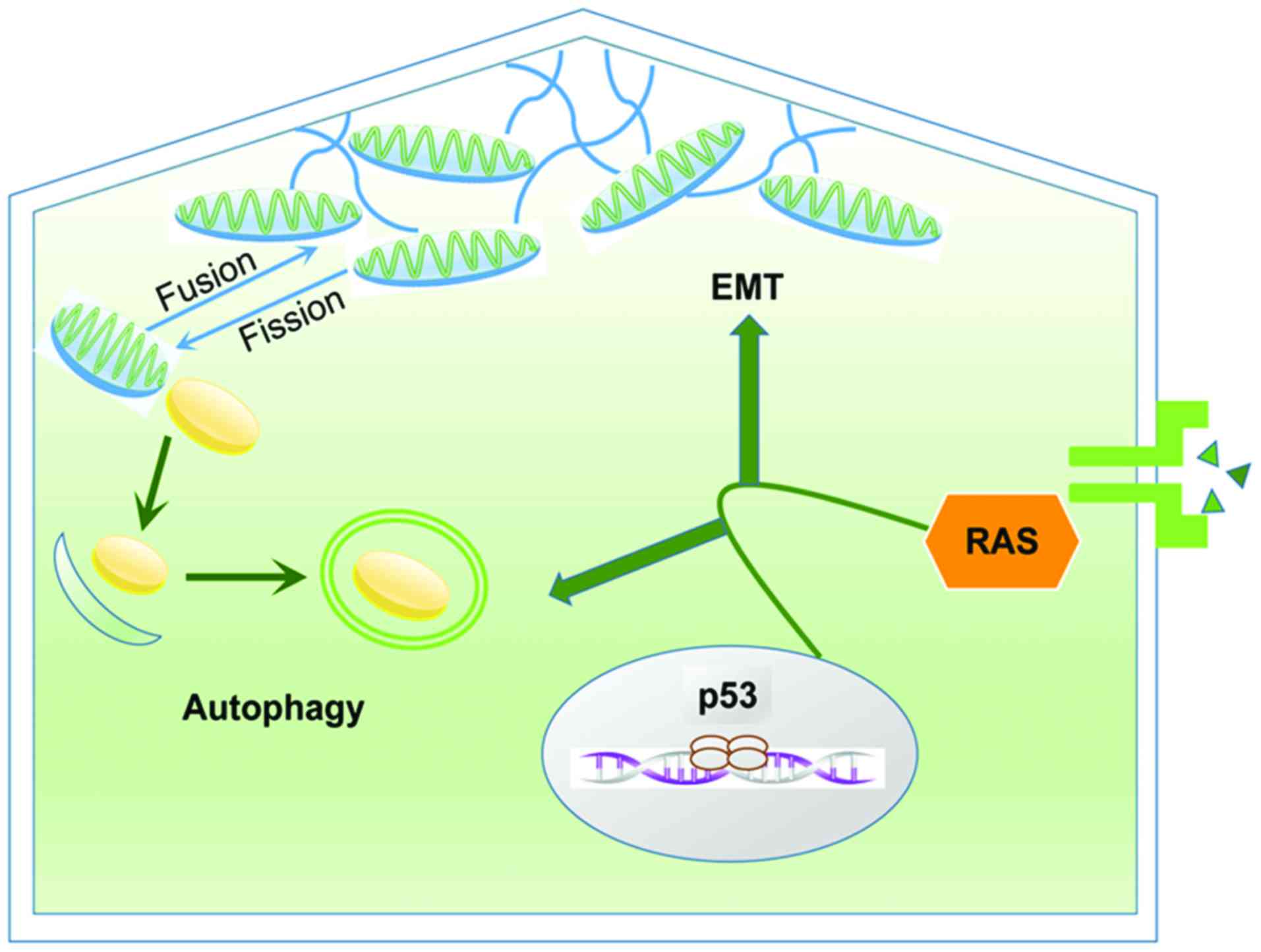
Нисимура Т., Тамура Н., Коно Н., Шиманака И., Араи Х., Ямамото Х. и др. Формирование аутофагосом инициируется в субдоменах ER, обогащенных фосфатидилинозитолсинтазой. EMBO J. 2017; 36 (12): 1719–35.
CAS PubMed PubMed Central Статья Google Scholar
Андреева Г., Гован С., Лин Дж., Фонг A-CLWT, Шамсаей Е., Паркс Х. Г. и др. Синтез фосфатидилхолина de novo необходим для образования и поддержания мембраны аутофагосом во время аутофагии. Аутофагия. 2020; 16 (6): 1044–60.
CAS PubMed Статья PubMed Central Google Scholar
Schütter M, Giavalisco P, Brodesser S, Graef M. Локальный канал жирных кислот в синтезе фосфолипидов стимулирует экспансию фагофора во время аутофагии.Клетка. 2020; 180 (1): 135–149.e14.
PubMed Статья CAS PubMed Central Google Scholar
Morishita H, Zhao YG, Tamura N, Nishimura T., Kanda Y, Sakamaki Y, et al. Критическая роль VMP1 в секреции липопротеинов. Малхотра В., Пфеффер С.Р., Пфеффер С.Р., Принц В.А., редакторы. eLife. 2019; 8: e48834.
CAS PubMed PubMed Central Статья Google Scholar
Tábara L-C, Escalante R. VMP1 устанавливает ER-микродомены, которые регулируют сайты контакта с мембраной и аутофагию. PLoS ONE. 2016; 11 (11): e0166499.
PubMed PubMed Central Статья CAS Google Scholar
Tábara L-C, Vicente J-J, Biazik J, Eskelinen E-L, Vincent O, Escalante R. Мембранный белок вакуоля 1 маркирует субдомены эндоплазматического ретикулума, обогащенные ферментами, синтезирующими фосфолипиды, и необходим для распределения фосфоинозидов.Движение. 2018; 19 (8): 624–38.
PubMed Статья CAS PubMed Central Google Scholar
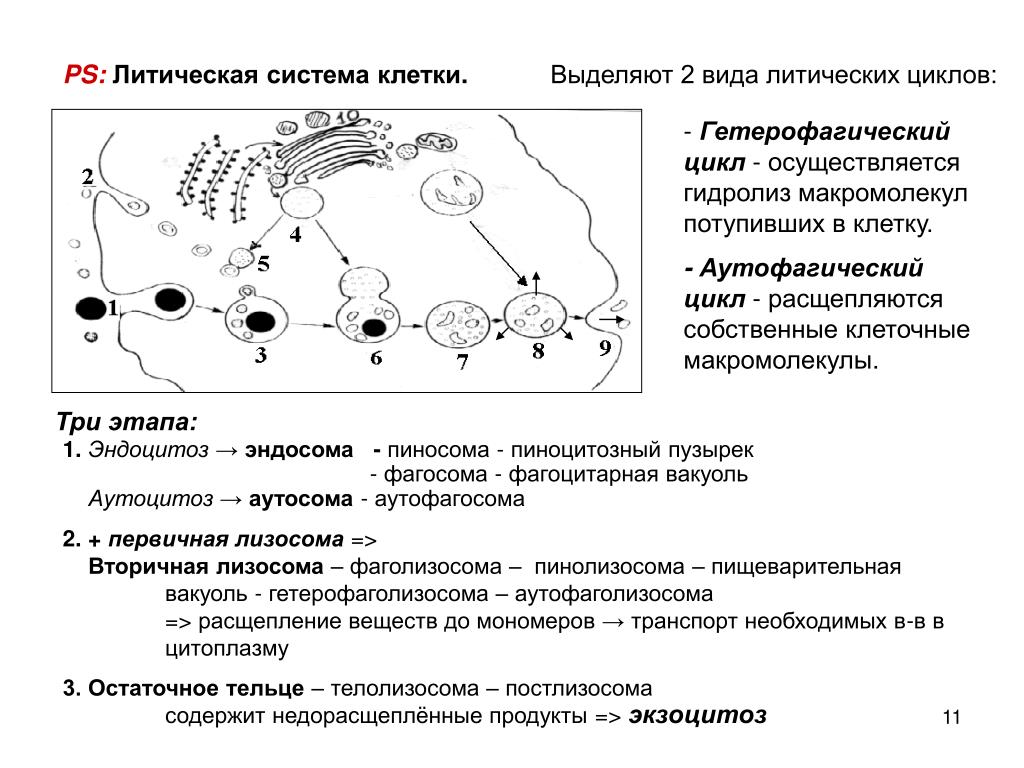
Zhao YG, Chen Y, Miao G, Zhao H, Qu W, Li D, et al. ER-локализованный трансмембранный белок EPG-3 / VMP1 регулирует активность SERCA, чтобы контролировать изоляцию ER-мембранных контактов для образования аутофагосом. Mol Cell. 2017; 67 (6): 974–989.e6.
CAS PubMed Статья PubMed Central Google Scholar
Морита К., Хама Ю., Идзуме Т., Тамура Н., Уэно Т., Ямасита Ю. и др. Полногеномный CRISPR-скрининг идентифицирует TMEM41B как ген, необходимый для образования аутофагосом. J Cell Biol. 2018; 217 (11): 3817–28.
CAS PubMed PubMed Central Статья Google Scholar
Моретти Ф., Бергман П., Доджсон С., Марселлин Д., Клаерр И., Гудвин Дж. М. и др. TMEM41B — новый регулятор аутофагии и мобилизации липидов. EMBO Rep.2018; 19 (9): e45889.
PubMed PubMed Central Статья CAS Google Scholar
Шима Т., Кирисако Х., Накатогава Х. Везикулы COPII вносят вклад в аутофагосомные мембраны. J Cell Biol. 2019; 218 (5): 1503–10.
CAS PubMed PubMed Central Статья Google Scholar
Нода Т. Аутофагия в контексте клеточной мембранно-транспортной системы: загадка пузырьков Atg9.Biochem Soc Trans. 2017; 45 (6): 1323–31.
CAS PubMed PubMed Central Статья Google Scholar
Ямамото Х., Какута С., Ватанабэ ТМ, Китамура А., Секито Т., Кондо-Какута С. и др. Везикулы Atg9 являются важным мембранным источником на ранних стадиях образования аутофагосом. J Cell Biol. 2012. 198 (2): 219–33.
CAS PubMed PubMed Central Статья Google Scholar
Puri C, Manni MM, Vicinanza M, Hilcenko C, Zhu Y, Runwal G, et al. Мутация центроядерной миопатии DNM2 выявляет связь между рециклингом расщепления эндосом и аутофагией.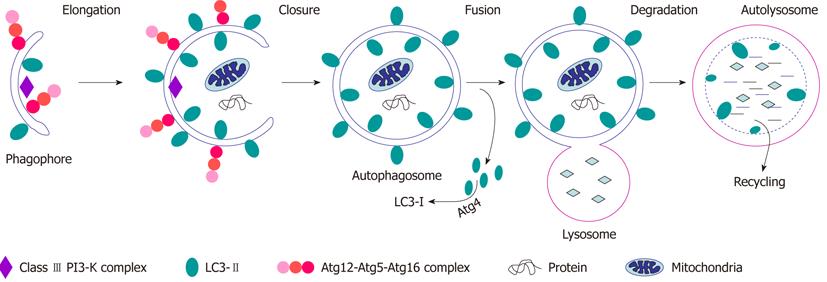 Dev Cell. 2020; 53 (2): 154–168.e6.
Dev Cell. 2020; 53 (2): 154–168.e6.
CAS PubMed Статья Google Scholar
Kampinga HH, Craig EA. Механизм шаперона HSP70: J-белки как драйверы функциональной специфичности. Nat Rev Mol Cell Biol. 2010. 11 (8): 579–92.
CAS PubMed PubMed Central Статья Google Scholar
Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 необходим в качестве дисульфидредуктазы для деградации неправильно свернутых белков в ER. Наука. 2008. 321 (5888): 569–72.
CAS PubMed Статья Google Scholar
Ямамото Ю., Кимура Т., Момохара С., Такеучи М., Тани Т., Кимата Ю. и др. Новый ER J-белок DNAJB12 ускоряет ER-ассоциированную деградацию мембранных белков, включая CFTR. Функция сотовой структуры.2010. 35 (2): 107–16.
CAS PubMed Статья Google Scholar
Медицинское определение аутофагосомы | Медицинский словарь Merriam-Webster
au · to · phago · некоторые | \ ˌȮ-tō-ˈfag-ə-ˌsōm \ : двойная мембраносвязанная везикула, которая включает в себя клеточные компоненты и сливается с лизосомами, которые переваривают эти клеточные компоненты во время аутофагии.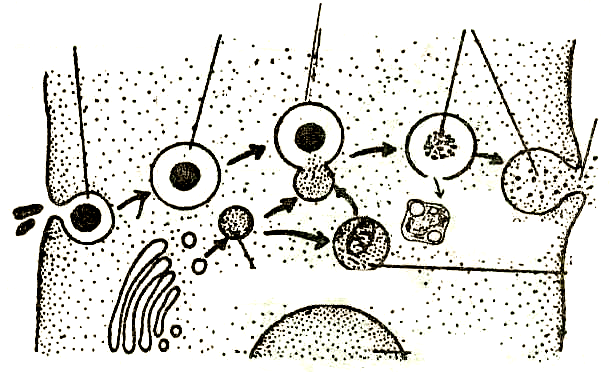 Аутофагосома не отделяется от ранее существовавших органелл, а, скорее, формируется в результате динамического процесса расширения мембраны.- Дэниел Дж. Клионски, Медицинский журнал Новой Англии , 23 апреля 2009 г. Во время аутофагии часть цитоплазмы изолируется в аутофагосому : затем она сливается с лизосомой, и материалы, полученные из цитоплазмы, разрушаются. — Сатоши Цукамото и др., Science , 4 июля 2008 г.
Аутофагосома не отделяется от ранее существовавших органелл, а, скорее, формируется в результате динамического процесса расширения мембраны.- Дэниел Дж. Клионски, Медицинский журнал Новой Англии , 23 апреля 2009 г. Во время аутофагии часть цитоплазмы изолируется в аутофагосому : затем она сливается с лизосомой, и материалы, полученные из цитоплазмы, разрушаются. — Сатоши Цукамото и др., Science , 4 июля 2008 г.Происхождение мембраны аутофагосом у млекопитающих
Аутофагия начинается с зарождения фагофоров, которые затем расширяются, давая начало двухмембранным аутофагосомам.Аутофагосомы в конечном итоге сливаются с лизосомами, где цитозольные грузы разрушаются. Накопление аутофагосом является признаком аутофагии и нейродегенеративных расстройств, включая болезнь Альцгеймера и Хантингтона. В последние годы источники мембран аутофагосом вызвали большой интерес, даже при этом доноры мембран для аутофагосом все еще остаются предметом обсуждения. В этом обзоре мы описываем возможные источники мембраны аутофагосомы.
1. Введение
Макроаутофагия (далее известная как аутофагия) — это неселективный процесс «самопоедания», который поддерживает клеточный гомеостаз, управляет реакциями на стресс и контролирует качество крупных белков и цитоплазматических компонентов путем устранения дефектных или избыточных молекул / структур, таких как неправильно свернутые белки, поврежденные митохондрии, избыточные пероксисомы, рибосомы и вторгшиеся патогены.Аутофагия, которая может быть вызвана экзогенными стимуляциями, такими как голодание питательными веществами, стресс эндоплазматического ретикулума (ЭР), лечение рапамицином, витамином D3 и IFN- γ , обеспечивает источник питания и экстренной реакции в периоды стресса для поддержания здоровья клеток гомеостаз и синаптическая функция. Дисфункция аутофагии — это неправильно регулируемый процесс при различных нейродегенеративных заболеваниях, различных типах рака, аутоиммунных заболеваниях и неконтролируемых инфекциях, характеризующихся накоплением белковых агрегатов, деградацией внутриклеточных патогенов и удалением стареющих органелл, включая болезнь Альцгеймера, болезнь Хантингтона, болезнь Паркинсона. болезнь и боковой амиотрофический склероз.
Функция аутофагии зависит от образования двойных или многомембранных везикул, называемых аутофагосомами (AP), которые играют ключевую роль в гомеостазе клетки, поглощая грузы, включая поврежденные митохондрии (митофагия) и белковые агрегаты. Активность аутофагии в первую очередь модулируется размером и количеством AP. Производство / накопление AP, которые впоследствии не соединяются с лизосомами (или накопление AP), напрямую вызывает клеточную токсичность в условиях различных стрессовых состояний, таких как окисление и агрегация токсичных белков, и этот процесс может быть вовлечен в патогенез нейродегенеративных заболеваний, туморогенез. и инфекции, среди прочего.Кислый pH и ферментативное действие гидролаз в лизосомах приводят к разрушению внутренних мембран AP, а также их содержимого. Однако до сих пор происхождение аутофагосомальной мембраны и молекулярные механизмы образования AP все еще неизвестны.
Однако до сих пор происхождение аутофагосомальной мембраны и молекулярные механизмы образования AP все еще неизвестны.
Здесь мы систематизируем и резюмируем статьи в этом выпуске по четырем основным направлениям: морфология, образование, функция и источник AP.
2. Морфология аутофагосомы
Будучи двух- или многомембранной органеллой, AP в цитоплазме является отличительной чертой и ключевым исходным событием аутофагии.Иногда двухмембранную структуру, которая содержит часть цитоплазматических компонентов, также можно расценить как AP. Размер и количество AP могут отдельно регулироваться различными подгруппами белков генов аутофагии (ATG) и членами семейства белков ATG8 / легкой цепи 3 (LC3). Это имеет смысл, учитывая, что изменение размера AP может влиять в первую очередь на селективность груза, в то время как регулирование количества AP может быть выполнено для регулирования в основном потока аутофагии. У млекопитающих время образования ПД составляет 5-10 мин [1].График голода определяет степень расширения AP. Количество белка ATG9 коррелирует с количеством аутофагических телец; то есть уровни ATG9 определяют количество AP. Азотное голодание индуцирует AP размером от 300 до 900 нм в диаметре, что больше, чем у большинства других везикул в клетке. Во время неселективной аутофагии преждевременное закрытие фагофора приводит к уменьшению АД.
Другое исследование показало, что размер AP может также зависеть от конкретного груза [2], который может варьироваться от белков до внутриклеточных бактерий (0.06 до 0,2 мкм м) [3]. Размер и образование AP регулируются стационарным уровнем ассоциированного с микротрубочками белка LC3 [4], но регуляторный механизм этого белка не изучен. Размер AP, вероятно, определяется отдельными аутофагическими шагами. Во-первых, AP могут расширяться за счет добавления мембраны с образованием изолирующих мембран на ранней стадии аутофагии. Во-вторых, AP могут расти путем слияния с эндосомами и лизосомами на поздней стадии аутофагии, хотя слияния эндосом и лизосом недостаточно для правильного роста аутофагосом. Кроме того, степень расширения AP в основном зависит от продолжительности условий индукции, таких как голодание. Снижение клеточного уровня ATG8, который закреплен на поверхности AP, приводит к меньшему AP по сравнению со штаммом дикого типа, но количество AP такое же, как и у AP дикого типа. Между тем, AP представляет собой высокодинамичную органеллу, и ее протеом отличается, если клетки находятся в условиях голодания или базальной макроаутофагии при блокировании конканамицином A.
Кроме того, степень расширения AP в основном зависит от продолжительности условий индукции, таких как голодание. Снижение клеточного уровня ATG8, который закреплен на поверхности AP, приводит к меньшему AP по сравнению со штаммом дикого типа, но количество AP такое же, как и у AP дикого типа. Между тем, AP представляет собой высокодинамичную органеллу, и ее протеом отличается, если клетки находятся в условиях голодания или базальной макроаутофагии при блокировании конканамицином A.
3.Формирование аутофагосомы
AP — это сложная серия дискретных событий, которые опосредуются и контролируются большим количеством белков, но этот процесс плохо изучен. По сути, AP образуются в результате индукции, экспансии (фагофор / изоляционная мембрана и омегасома), завершения везикул (AP), слияния (амфисома) и деградации (аутолизосома / лизосома) [5], как показано на рисунке 1. В головном мозге биогенез ПД происходит дистально в конститутивном процессе на кончике нейрита, который находится далеко от ядра, в зависимости от моторного комплекса микротрубочек и динеин-динактина.Различные стрессовые условия, такие как голодание, окисление и агрегация токсичных белков, могут ускорить биогенез AP и деградацию лизосом. Зрелый AP генерируется, когда изолирующая мембрана закрывается, а затем зрелые AP сливаются с вакуолью / лизосомой, где содержимое разлагается, а продукты рециркулируют в цитозоль для повторного использования. Идентифицировано около 40 ATG, и большинство из них консервативны у высших эукариот, но только части ATG непосредственно связаны с биогенезом AP млекопитающих, как показано в таблице 1.Гомотипическое слияние ATG16-положительных и LC3-отрицательных предшественников AP является критическим регуляторным этапом в биогенезе AP. Как конъюгированная форма LC3, LC3-II связан с внешней частью аутофагосомных мембран после завершения и остается с AP до слияния с лизосомами [6]. Кроме того, повышенные уровни LC3-II обычно коррелируют с накоплением AP в клетке, но, таким образом, не указывают на увеличение биогенеза AP.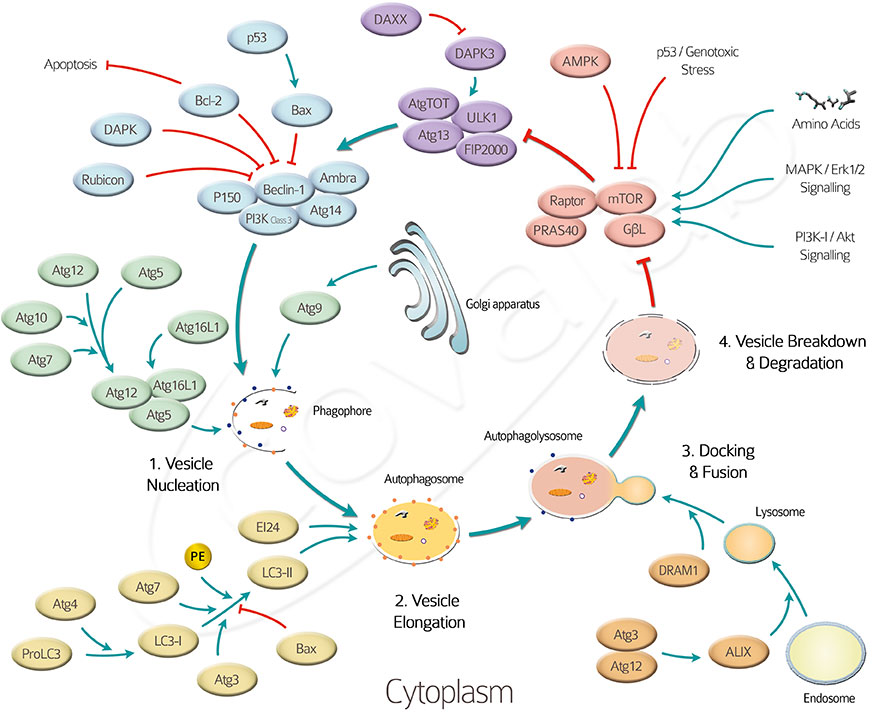
905 905 905 ] 90535 TORC24351 905in требуется сортировка ne для эффективной аутофагии и мембранных канальцев35 205 белок Atg -Atg31-Atg17 комплекс [21], образование димера с двумя полумесяцами для слияния в расширяющийся фагофор
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
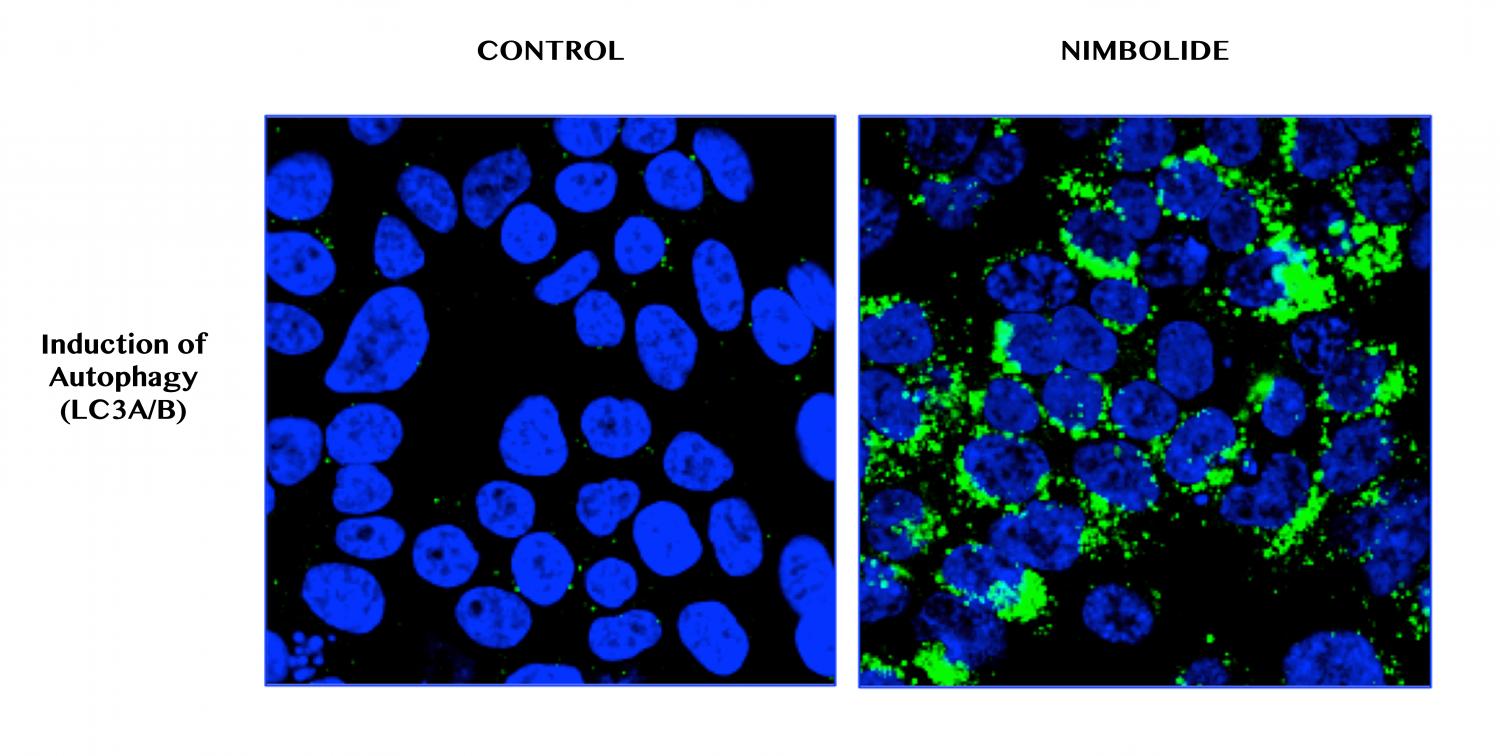
Есть сообщения, что ULK1 / ATG1-ATG Были обнаружены протеинкиназный комплекс, комплекс VPS (вакуумная сортировка белков) 34 (VPS34), система транспортировки ATG9, комплекс ATG5-ATG12-ATG16L1, два убиквитиноподобных белка ATG12 и ATG8 / LC3 и их системы конъюгации, которые приводят к образованию и расширению фагофора, который в конечном итоге закрывается, образуя полный AP.Между тем, образование AP сильно индуцируется. Аминокислота может вызывать аутофагию, а комплекс протеинкиназ TORC1 / mTORC1 подавляет образование AP в условиях, богатых питательными веществами. Белок актина также необходим для опосредованной голоданием аутофагии через комплекс Arp2 / 3, а WHAMM и деполимеризация актина участвует в образовании AP на очень ранней стадии, а не на стадиях созревания. Кроме того, деконъюгация ATG8-phosphatidylethanolamine (PE) также необходима для эффективного биогенеза AP для оптимальной экспансии фагофоров.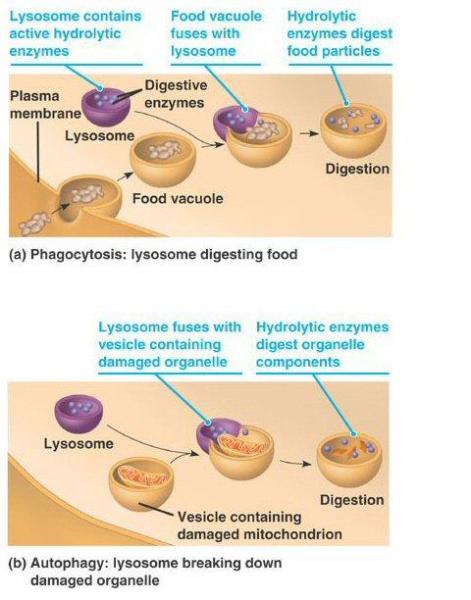
4. Функция аутофагосомы
Аутофагия — это эволюционно законсервированный клеточный процесс для поддержания энергетического гомеостаза. Как маркер аутофагии, динамика и функции AP остаются устойчивыми в мышиной модели нейродегенеративного заболевания, но поток AP не увеличивается, даже когда белок накапливается вдоль аксона [23]. В настоящее время стимуляция синтеза AP часто используется для усиления аутофагии, чтобы снизить токсичность агрегации белка при нейродегенерации и старении. Ретроградный транспорт AP может играть роль в процессах передачи нейрональных сигналов, способствуя морфологической сложности нейронов и предотвращая нейродегенерацию.Аутофагия может способствовать заражению пикорнавирусами, такими как полиовирус и вирусы Коксаки, только потому, что AP предоставляют сайты для репликации. Как и везикулы с двойной мембраной, состав внешней и внутренней мембран AP кажется совершенно различным [24]. Итак, разные мембраны выполняют разные роли: внутренняя аутофагосомная мембрана отвечает за секвестрацию грузов, а внешняя аутофагосомная мембрана отвечает за слияние с лизосомальной мембраной.
Между тем две характеристики делают точки доступа уникальным типом сотовой транспортной сети.Во-первых, груз окружают два липидных бислоя, а во-вторых, эти гигантские везикулы обычно имеют средний диаметр примерно 700 нм, которые могут дополнительно расширяться, чтобы вместить большие структуры, такие как клеточные органеллы и бактерии [25]. Но накопление AP вызывает цитотоксичность; особенно в определенных стрессовых условиях избыточные AP, впоследствии не слившиеся с лизосомами, напрямую вызывают клеточную токсичность независимо от апоптоза и некроптоза, и этот процесс может означать, что AP является признаком нейродегенеративных расстройств, включая болезнь Альцгеймера и Хантингтона или боковой амиотрофический склероз [26, 27]. .
5. Источник мембраны аутофагосомы
Поскольку аутофагия — это неселективный путь массовой деградации, специфическое мембранное происхождение всех AP остается неясным, хотя морфологические особенности AP в основном являются общими для традиционной и альтернативной аутофагии. В отличие от дрожжевых и растительных клеток, в клетках млекопитающих отсутствует преаутофагосомная структура (PAS). Предполагается, что участки выхода эндоплазматического ретикулума (ERES), митохондрии, участки контакта ER-митохондрий, промежуточный компартмент ER-Golgi (ERGIC), аппарат Гольджи и плазматическая мембрана (PM) доставляют липиды к растущей изоляционной мембране в клетках млекопитающих. но точный механизм, опосредующий этот процесс, остается неясным.Возможно, что существуют разные источники мембран в зависимости от типа клеток, условий роста, физиологических условий и т. Д., Как показано в таблице 2.
В отличие от дрожжевых и растительных клеток, в клетках млекопитающих отсутствует преаутофагосомная структура (PAS). Предполагается, что участки выхода эндоплазматического ретикулума (ERES), митохондрии, участки контакта ER-митохондрий, промежуточный компартмент ER-Golgi (ERGIC), аппарат Гольджи и плазматическая мембрана (PM) доставляют липиды к растущей изоляционной мембране в клетках млекопитающих. но точный механизм, опосредующий этот процесс, остается неясным.Возможно, что существуют разные источники мембран в зависимости от типа клеток, условий роста, физиологических условий и т. Д., Как показано в таблице 2.
905 37] (MAM) 905 23905 905 мембранные липиды
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.
 Сайты контакта ER-митохондрий [28]
Сайты контакта ER-митохондрий [28]Сайты контакта ER-митохондрий в последнее время привлекают большое внимание. В 1952 году Вильгельм Бернхард впервые сообщил о сайтах контакта ER-митохондрий в печени крыс с помощью электронных микрофотографий [29]. Сайты контакта ER-митохондрий важны для нескольких ключевых клеточных процессов, таких как гомеостаз кальция, метаболизм липидов, регуляция аутофагии, митохондриальная динамика [30], выживаемость клеток для энергетического метаболизма и сворачивание белков [31]. Правильное поддержание интерфейса ER-митохондрии является важной частью аутофагического процесса; Между тем появление фагофоров и места контакта между ЭР и митохондриями обнаруживают удивительную корреляцию.После голодания все компоненты PI3K класса III, специфичного для аутофагии (ATG14L, Vps34, Beclin1 и Vps15), накапливаются во фракции митохондриально-ассоциированных мембран (MAMs) и, вероятно, рекрутируются в сайты контакта ER-митохондрий после индукции аутофагии, и аутофагосома -формационный маркер ATG5 также локализуется в сайте контакта ER-митохондрий до завершения образования AP. А образование AP значительно подавляется в клетках с нокаутом митофузина 2, в которых нарушены сайты контакта ER-митохондрий [32].
7. Места выхода эндоплазматического ретикулума
Как самая большая органелла в клетке, ER находится в непосредственной близости с другими компартментами эндомембраны из-за синтеза липидов и белков мембран и их наружного транспорта, устанавливая участки мембранно-мембранного контакта (MCS), которые облегчают сигнальные события, модулируют динамические процессы органелл и обменивают липиды и ионы. ERES, специализированные области ER, где генерируются транспортные пузырьки COPII, как полагают, пространственно, физически и функционально связаны с AP.Ранние аутофагические структуры тесно связаны с мембраной ER из-за присутствия субдомена омегасом, положительного по двойному FYVE-содержащему белку 1 (DFCP1), фосфатидилинозитол-3-фосфат (PI3P) связывающему белку; между тем, изоляционная мембрана омегасом и AP фагофор, вероятно, все происходят из ER [34]. Sandra Maday [45] также считает, что APs генерируются в DFCP1-позитивных субдоменах ER на дистальном конце аксона, отличных от сайтов выхода ER в первичных нейронах.Исследования показали, что [46] ассоциированный с мембраной лизосом белок-2 (LAMP-2), как сильно гликозилированный мембранный белок типа 1, должен иметь решающее значение для транслокации синтаксина-17 (STX17) на мембраны аутофагосом. Sanchez-Wandelmer et al. [47] обнаружили, что ERES являются ключевыми элементами в формировании изолирующих мембран и ассоциатов ER с расширением изолирующих мембран или фагофоров в клетках млекопитающих с помощью электронной томографии. Однако появились противоречивые данные, указывающие на то, что только 30% всех AP связаны с ER или специализированными областями ER [36].
Sandra Maday [45] также считает, что APs генерируются в DFCP1-позитивных субдоменах ER на дистальном конце аксона, отличных от сайтов выхода ER в первичных нейронах.Исследования показали, что [46] ассоциированный с мембраной лизосом белок-2 (LAMP-2), как сильно гликозилированный мембранный белок типа 1, должен иметь решающее значение для транслокации синтаксина-17 (STX17) на мембраны аутофагосом. Sanchez-Wandelmer et al. [47] обнаружили, что ERES являются ключевыми элементами в формировании изолирующих мембран и ассоциатов ER с расширением изолирующих мембран или фагофоров в клетках млекопитающих с помощью электронной томографии. Однако появились противоречивые данные, указывающие на то, что только 30% всех AP связаны с ER или специализированными областями ER [36].
8. Митохондрии
Митохондрии являются наиболее важными органеллами, определяющими фундаментальную метаболическую активность, гомеостаз железа и кальция и передачу сигналов различными клеточными путями. Дисфункция и нарушение регуляции митохондрий могут привести ко многим заболеваниям человека, включая сердечно-сосудистые заболевания, нейродегенеративные заболевания и рак. Ряд сообщений предполагает, что [32] существует связь между липидами внешней мембраны митохондрий, белками и образованием AP.Предполагается, что внешняя митохондриальная мембрана передает поток липидов и мембранных белков AP. Белок ранней аутофагии ATG5 и аутофагосомный маркер LC3 перемещаются в точки, локализованные на внешней мембране митохондрий после голодания, подтверждая, что митохондрии играют центральную роль в аутофагии, вызванной голоданием. Между тем, исследования обнаружили донорство митохондриальной мембраны для образования AP как при базальной (в присутствии сыворотки и носителя), так и при индуцированной лекарством аутофагии в клеточной линии рака груди человека, за исключением захвата формирующимся AP [48].В других исследованиях авторы также показывают, что связь между ER и митохондриями имеет решающее значение, поскольку в ее отсутствие APs, вызванные голоданием, не образуются [32]. Контраргумент состоит в том, что митохондрии не имеют ничего общего с источником AP-мембран, поскольку обнаружено, что ATG9 млекопитающих локализуется только в сети транс-Гольджи (TGN) и поздних эндосомах, но не в митохондриях [49], в то время как компартменты, содержащие ATG9, являются источник мембран для образования и / или расширения ПД.
Контраргумент состоит в том, что митохондрии не имеют ничего общего с источником AP-мембран, поскольку обнаружено, что ATG9 млекопитающих локализуется только в сети транс-Гольджи (TGN) и поздних эндосомах, но не в митохондриях [49], в то время как компартменты, содержащие ATG9, являются источник мембран для образования и / или расширения ПД.
9.Аппарат Гольджи
Аппарат Гольджи является основным сайтом гликозилирования, участвующим в синтезе, модификации и секреции белков и липидов. Предполагается, что аппарат Гольджи является основным источником мембран для образования AP млекопитающих. Этот результат был впервые обнаружен Локком и Коллинзом в 1965 году при разработке клеток жирового тела беспозвоночных. Другие исследования показали, что комплекс Гольджи способствует ранней стадии аутофагии [50]. В телофазе клетки снова наблюдается распределение структур Гольджи в APs по периферии клетки, когда известно, что аппарат Гольджи собирается заново.На основании этого они предположили, что аппарат Гольджи является мембранным источником для роста аутофагосом. Как единственный трансмембранный белок ATG, ATG9 ассоциирован с аппаратом Гольджи и может участвовать в обеспечении мембраны для образования AP [51]. Однако др. Утверждали, что ATG9 млекопитающих (mATG9 и ATG9L1) ассоциируется со многими др. Компартментами, включая рециклирующие эндосомы, ранние эндосомы и поздние эндосомы [37]. Возможно, что все эти органеллы участвуют в образовании AP.
10. Плазменная мембрана
PM, который образует барьер между цитоплазмой и окружающей средой [52], играет критическую роль в повышении вирулентности, опосредуя секрецию факторов вирулентности, эндоцитоз, синтез клеточной стенки и инвазивный морфогенез гиф. Между тем, белки в PM также опосредуют транспорт питательных веществ и определяют pH, осмолярность, питательные вещества и другие факторы во внеклеточной среде. Способность PM вносить вклад в образование AP может быть особенно важной во время увеличения аутофагии в клетках млекопитающих.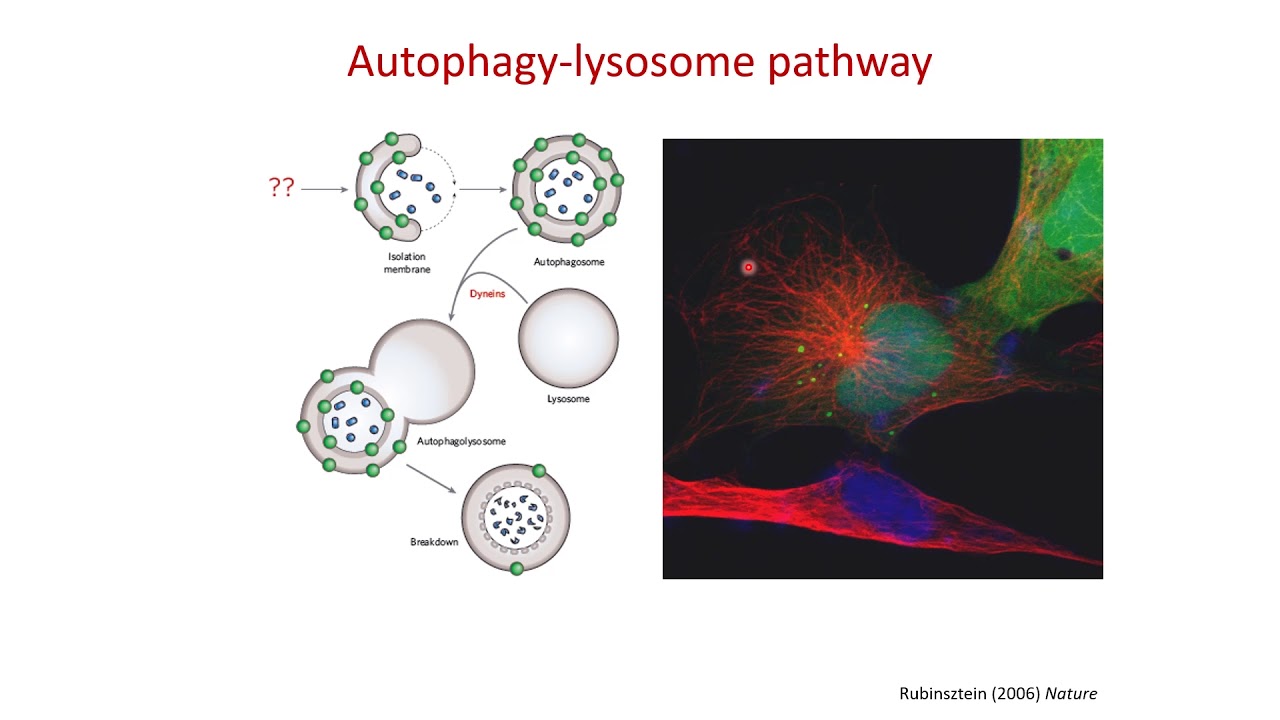 Большая площадь поверхности PM может действовать как массивный мембранный накопитель, который позволяет клеткам испытывать циклы синтеза AP с гораздо более высокой скоростью, чем в основных условиях, без ущерба для других процессов. ATG и мембраны, необходимые для образования AP, происходят из PM [47]. Клаудиа Пури и др. обнаружили, что ATG16L1 ассоциирует с ямками, покрытыми клатрином, и после интернализации и снятия оболочки, ATG16L1-ассоциированный PM становится ассоциированным с предшественниками фагофоров, которые созревают в фагофоры, а затем в APs [53].
Большая площадь поверхности PM может действовать как массивный мембранный накопитель, который позволяет клеткам испытывать циклы синтеза AP с гораздо более высокой скоростью, чем в основных условиях, без ущерба для других процессов. ATG и мембраны, необходимые для образования AP, происходят из PM [47]. Клаудиа Пури и др. обнаружили, что ATG16L1 ассоциирует с ямками, покрытыми клатрином, и после интернализации и снятия оболочки, ATG16L1-ассоциированный PM становится ассоциированным с предшественниками фагофоров, которые созревают в фагофоры, а затем в APs [53].
11. Рециркуляция эндосом
В клетках млекопитающих эндосомная система чрезвычайно динамична и генерирует несколько структурно и функционально различных компартментов, а именно ранние / рециркулирующие эндосомы (RE), поздние эндосомы и лизосомы. Идентичность эндосом обеспечивается специфической локализацией регуляторов. RE состоят из трубчатой сети, которая выходит из вакуолярных сортирующих эндосом и направляет грузы к поверхности клетки, Гольджи или органеллам, связанным с лизосомами.RE также участвуют в образовании AP. mATG9, передаваемый от PM к RE, необходим для инициации и развития аутофагии. A. Orsi и его исследовательская группа обнаружили, что mATG9-позитивные структуры динамически взаимодействуют с фагофорами и AP. TBC1D14, белок домена Tre-2 / Bub2 / Cdc16 (TBC), регулирует RE, положительные по рецептору Tfn (TfnR-), которые необходимы для образования AP. Как положительный регулятор образования AP, мембранный ремоделирующий белок, сортирующий nexin18 (SNX18), необходим для регуляции транспортировки ATG9A из RE и образования мембран-предшественников ATG16L1 и WIPI2-позитивных AP [54].ATG16L1 и mATG9-положительные везикулы присутствуют в одних и тех же сайтах на RE, что может снижать выход мембраны из RE, чтобы увеличить образование AP. Все это указывает на то, что RE, вероятно, являются донорами мембран для SNX18-обеспечиваемого биогенеза AP.
12. Промежуточный отсек эндоплазматической сети-Гольджи
Структуры ERGIC могут перемещаться от ERES к аппарату Гольджи путем отслеживания на микротрубочках. Мембранный трафик между ER и Golgi является двунаправленным и происходит с помощью механизмов, аналогичных другим MAM.Недавно ERGIC, мембранный отсек между ER и Golgi для сортировки и переработки грузов, был предложен в качестве еще одного мембранного источника для фагофора. На интерфейсе ER-Golgi везикулы комплекса белков оболочки I (COPI) зачатки и облегчают ретроградный транспорт от Golgi и ERGIC, тогда как везикулы COPII могут быть предшественниками мембраны фагофора. В качестве донорской мембраны ERGIC представляет собой сортировочную станцию, претерпевающую динамический мембранный обмен с везикулами COPI и COPII [55], последние из которых предположительно являются источником мембран для AP в ERGIC.Liang Ge et al. [44] также обнаружили, что генерация везикул COPII из ERGIC может быть особым событием для связанной с аутофагией мобилизации мембран, индуцированной ремоделированием ERES, вызванным голоданием. Лекарства, нарушающие ERGIC, также подавляли конъюгацию LC3 и образование точек LC3.
13. Контактные участки мембран ER-плазмы (ER-PMcs)
ER-PMcs также мобилизуются для биогенеза AP. В настоящее время исследования идентифицировали некоторые функции ER-PMcs, например, регулируют передачу сигналов Ca2 + [56] и сохраняют гомеостаз липидов.Исследовательская группа Nascimbeni AC показала, что ER-PMcs могут связываться с белками расширенных синаптотагминов (E-Syts), которые необходимы для синтеза фосфатидилинозитол-3-фосфата (PI3P), связанного с аутофагией, на кортикальной мембране ER, а затем регулировать биогенез АП млекопитающих [57].
Согласно другой литературе, цитоплазматические везикулы, содержащие ATG9 (везикулы ATG9) [58], контактный сайт ER-липидной капли (LD) [59] и везикулы COPII [11] также считаются источниками мембран для создания AP .Третьи думают, что ассоциированные с ATG9 мембраны не сливаются с APs и регулируют аутофагию только из-за своих временных структурных или каталитических функций [60].
14. Заключение
Снижение накопления AP может быть вариантом лечения нейродегенеративных заболеваний с помощью белковых агрегатов, поэтому убедитесь, откуда берется AP-мембрана и как она образуется? В последние годы эти вопросы вызывают большой интерес. ER, митохондрии, комплекс Гольджи и плазматическая мембрана были предложены в качестве источников аутофагосомных мембран.Конечно, происхождение AP-мембраны — это мультиисточники вероятности, что подтверждается все большим количеством исследований. Различное происхождение AP мембраны взаимодействует друг с другом. Эти разные выводы, сделанные разными лабораториями, могут быть частично связаны с разными экспериментальными подходами и методами, используемыми в разных лабораториях. И относительный вклад каждого источника при любом одном наборе условий еще предстоит определить. Хотя были предложены различные источники AP-мембран, неясно, в какой степени они могут быть взаимоисключающими или могут ли они сливаться и сотрудничать.Требуется много исследований.
ER, митохондрии, комплекс Гольджи и плазматическая мембрана были предложены в качестве источников аутофагосомных мембран.Конечно, происхождение AP-мембраны — это мультиисточники вероятности, что подтверждается все большим количеством исследований. Различное происхождение AP мембраны взаимодействует друг с другом. Эти разные выводы, сделанные разными лабораториями, могут быть частично связаны с разными экспериментальными подходами и методами, используемыми в разных лабораториях. И относительный вклад каждого источника при любом одном наборе условий еще предстоит определить. Хотя были предложены различные источники AP-мембран, неясно, в какой степени они могут быть взаимоисключающими или могут ли они сливаться и сотрудничать.Требуется много исследований.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Благодарности
Авторы выражают признательность за финансовую поддержку Пекинскому фонду естественных наук (№ 7162169) и Национальному фонду естественных наук Китая (№ 81173383, 81573819).
Начало конца
52
было исследовано на ограничивающих мембранах зарождающихся или вновь образованных аутофаго-
сомов в печени крыс, лишенных аминокислот (Dunn 1990).Белковые маркеры грубого ER
были обнаружены на этих промежуточных соединениях, в то время как маркеры Гольджи, плазматической мембраны,
и эндосом отсутствовали. Другие исследования, в которых мембраны печени крыс были фракционированы, показали, что фракции, обогащенные аутофагосомными мембранами, содержали
маркерных белков ER, но не белка Гольджи (Furuno et al. 1982; Kominami et al.
1983; Ueno et al. al. 1991). Это мнение позже было косвенно подтверждено наблюдением, что дрожжевые мутанты с дефектом COPII-опосредованного транспорта из ER dis-
нарушают аутофагию (Ishihara et al.2001), что также подтверждается исследованиями
инфицированных клеток. В частности, Listeria monocytogenes, содержащий компонент com-
, присутствующий в цитоплазме инфицированных макрофагов, который, по-видимому, имеет аутофагосомное происхождение
, является положительным в отношении грубого маркерного белка дисульфида
изомеразы (PDI) ER (Rich et al. 2003 г.). Более того, экспрессия белков полиовируса
в клетках COS-1 индуцировала образование двухмембранных везикул из ЭПР, которые морфологически напоминают аутофагосомы
(Suhy et al.2000).
Исследование двойного FYVE-домена-содержащего белка 1 (DFCP1), связывающего белок PtsIns3P-
, предоставило первые убедительные доказательства того, что ER может быть источником
для мембран аутофагосом. DFCP1 локализуется в ER и Golgi в питаемых клетках
и после аминокислотной депривации он перемещается в цитозольные точечные структуры
, которые являются LC3- и ATG5-положительными (Ax et al. 2008). Это перераспределение требует
прямого распознавания PtsIns3P, а также белков, таких как BECLIN 1 и PI3KC3
, составляющих специфический для аутофагии комплекс PtdIns3K, ответственный за продуцирование PtdIns3P
.Важно отметить, что исследования изображений живых клеток показали, что эти точечные структуры, образованные через 30 мин после индукции аутофагии, представляют собой кольцевые мембранные выступы
, окружающие аутофагосомный маркерный белок LC3. Поскольку они
часто наблюдаются в ассоциации с лежащим в основе ER, предполагая Ω-подобную конформацию, авторы назвали их омегасомами. Эксперименты по визуализации живых клеток
выявили тесную связь между омегасомами и возникающими аутофаго-
сом (Ax et al.2008 г.). На ранних стадиях формирования аутофагосомы,
небольшое количество DFCP1 концентрируется на краю цепи ER. По мере увеличения положительной области DFCP1-
LC3 начинает накапливаться в непосредственной близости от нее. Впоследствии омегасома расширяет и полностью окружает LC3-позитивную структуру предшественника цен-
трала до того, как LC3-позитивная аутофагосома выйдет из омега-
и весь DFCP1 реабсорбируется в ER.Недавно большее количество белков ATG
было локализовано в омегасомах (Matsunaga et al. 2010; Polson et al. 2010).
Вместе все эти данные указывают на то, что омегасомы являются местом, где формируются по крайней мере суб-
набор PAS и фагофоров (рис. 3.2).
Два исследования на основе электронной томографии подтвердили идею о том, что омега-
ом являются специализированными доменами ER, в которых образуются аутофагосомы. В первой работе
трехмерные реконструкции областей с формирующимися аутофагосомами
показали, что цистерны ER расположены параллельно фагофорам,
внутри и снаружи (Yla-Anttila et al.2009) (рис. 3.2). Кроме того, эти трехмерные проекции
показали, что цистерны ER внутри фагофора
проходят в цитоплазму через открытый конец фагофора. Кроме того, они
также показали, что ER и фагофорные мембраны связаны посредством
S. Abreu et al.
Примирование SNARE важно для созревания аутофагосом, но не для их образования
Значение
Вклад SNARE-опосредованного слияния мембран в различные стадии аутофагического процесса изучен недостаточно.В этой работе мы демонстрируем, что ингибирование прайминга SNARE приводит к нарушению аутофагического потока, что проявляется накоплением зрелых аутофагосом, которые не сливаются с лизосомами. Наши результаты показывают, что прайминг SNARE необходим для слияния аутофагосомы с лизосомой, но не для образования аутофагосом.
Abstract
Аутофагия, уникальный путь внутриклеточного мембранного переноса, инициируется образованием изолирующей мембраны (фагофор), которая поглощает компоненты цитоплазмы, что приводит к образованию аутофагосомы, везикулы с двойной мембраной, которая нацелена на лизосома.Следовательно, наружная мембрана аутофагосомы сливается с лизосомальной мембраной. Было высказано предположение, что множественные события слияния мембран, опосредованные молекулами SNARE, способствуют аутофагии. αSNAP, адапторная молекула для SNARE-примирующего фермента N -этилмалеимид-чувствительный фактор ( NSF ), как известно, имеет решающее значение для процессов слияния внутриклеточных мембран, но его роль в аутофагии остается неясной. Здесь мы продемонстрировали, что нокдаун αSNAP приводит к ингибированию аутофагии, что проявляется в накоплении запечатанных аутофагосом, расположенных в непосредственной близости от лизосом, но не слитых с ними.Более того, в этих условиях ассоциация как Atg9, так и синтаксина белка SNARE, связанного с аутофагией, с аутофагосомой осталась неизменной. Наконец, наши результаты показали, что в условиях голодания уровни αSNAP, хотя и низкие, тем не менее, достаточны для частичного стимулирования прайминга SNARE, необходимого для аутофагии. Взятые вместе, эти находки показывают, что хотя слияние аутофагосомно-лизосомальной мембраны чувствительно к ингибированию прайминга SNARE, начальные стадии биогенеза аутофагосом и экспансии аутофагосом остаются устойчивыми к его утрате.
Большинство событий слияния внутриклеточных мембран опосредуются молекулами растворимого рецептора белка прикрепления NSF (SNARE). Набор из трех Q / t-SNARE на целевой мембране и одного R / v-SNARE на транспортной везикуле создает комплекс SNARE-штырь, сближая мембрану и везикулу и, таким образом, обеспечивая их слияние (1, 2) . Как только эти две молекулы слились, отдельные молекулы SNARE высвобождаются из своего комплекса АТФ-зависимым образом с помощью фермента N -этилмалеимид-чувствительного фактора (NSF) и его адаптивного растворимого белка прикрепления NSF альфа (αSNAP) (3 ).Эти два белка являются универсальными компонентами клеточного транспорта и слияния, и удаление любого из них является летальным для клетки (4). Рекрутирование NSF АТФазы в комплекс SNARE полностью зависит от присутствия αSNAP. Три молекулы αSNAP направляют гомогексамерный NSF в белковый комплекс SNARE (5), стимулируя гидролиз АТФ и тем самым вызывая конформационные изменения в молекулах SNARE, которые приводят к их «праймированию» и позволяют им способствовать последующим событиям (6). Помимо αSNAP, существуют две дополнительные изоформы SNAP: βSNAP, который способствует слиянию мембран в головном мозге (7), и γSNAP, который, как недавно было показано, стимулирует разборку SNARE, особенно в эндосомах (8).Изменения внутриклеточных уровней αSNAP также вовлечены в различные патологические состояния, такие как нейродегенеративные заболевания, диабет и рак (см. Ссылку 9).
Аутофагия — это катаболический процесс, нацеленный на белки и органеллы для деградации в лизосомах. Он инициируется образованием изолирующей мембраны (также называемой фагофором), которая удлиняется и поглощает цитоплазматические компоненты с образованием аутофагосомы, везикулы с двойной мембраной, которая затем направляется к лизосоме.Эти последовательные стадии биогенеза аутофагосом требуют значительного ремоделирования мембран и процессов слияния мембран. Однако нет доказательств ретроградного транспорта из этого пузырька обратно к исходной мембране, это указывает на то, что праймирование SNARE может не быть важным для биогенеза аутофагосом.
Недавно сообщалось о роли молекул SNARE в аутофагическом процессе у дрожжей и в клетках млекопитающих. Комплексы SNARE, которые способствуют слиянию везикул с лизосомой у дрожжей, были идентифицированы как в пути цитоплазма-вакуоль (Cvt), так и в аутофагии, вызванной голоданием (10), а также в удлинении фагофора (11).В клетках млекопитающих и в Drosophila , молекулы SNARE были обнаружены важными для слияния аутофагосомно-лизосомальной мембраны (12, 13). Роль прайминга SNARE в биогенезе аутофагосом, однако, остается неясной. Первоначально сообщалось, что дрожжевые ортологи Sec18p и Sec17p, NSF и SNAP важны для слияния аутофагосомно-вакуолярной мембраны, но не для образования аутофагосом (14). Наир и др. (15), однако, недавно показали, что на формирование аутофагосом у дрожжей также могут влиять мутации в этих генах.В системе млекопитающих было высказано предположение, что молекулы SNARE необходимы для биогенеза аутофагосом; однако роль NSF и αSNAP в этой системе напрямую не рассматривалась (16, 17).
В настоящем исследовании мы использовали подход siRNA для изучения роли прайминга SNARE в аутофагии. Мы показали, что затруднение прайминга SNARE путем нокдауна αSNAP приводит к ингибированию аутофагии, сопровождающемуся накоплением аутофагосом, меченных LC3 и p62, в непосредственной близости от лизосом.Более того, наши результаты показали, что на перемещение синтаксина 17 (белка SNARE, связанного с аутофагией) из митохондрий в аутофагосомы не влияет снижение уровней αSNAP и не влияет на ассоциацию пузырьков Atg9 с аутофагической мембраной. Важно отметить, что голодные клетки были относительно более устойчивы к нокдауну αSNAP, возможно, из-за повышенной ассоциации молекул NSF с мембраной в местах, где это способствует праймингу SNARE. Таким образом, наши результаты подразумевают, что праймирование SNARE необходимо для производства аутофагосом, способных к слиянию с лизосомами, но они вполне необходимы для снабжения мембран, необходимого для образования аутофагосом.
Результаты
Прайминг SNARE необходим для аутофагического потока.
Прайминг SNARE, преимущественно опосредованный NSF и αSNAP, необходим для всех событий слияния внутриклеточных мембран. Однако его роль в аутофагии остается неясной. Пытаясь выяснить его роль, мы изучили влияние нокдауна αSNAP на аутофагическую активность клеток HeLa. Аутофагический поток контролировали в присутствии или в отсутствие лизосомального ингибитора бафиломицина А (BafA) с использованием как вестерн-блоттинга, так и иммунофлуоресцентного анализа.Истощение αSNAP привело к снижению аутофагического потока, на что указывает накопление LC3II (липидированная форма LC3, ортолога дрожжевого Atg8 у млекопитающих) и p62, хорошо охарактеризованного аутофагического груза (рис. 1 A и B ). Более того, в отличие от перинуклеарного распределения меченых везикул в контрольных клетках, нокдаун αSNAP приводил к диспергированию этих меченых везикул по всей цитоплазме (Fig. 1 B ). Аналогичные результаты были получены при использовании GATE-16, другого члена семейства белков mAtg8, для мониторинга аутофагии (рис.S1 A и B ). Следует отметить, что нокдаун NSF влияет на аутофагический поток еще в меньшей степени по сравнению с контрольной siRNA (Fig. S1 E и F ). Кроме того, экспрессия доминантно-отрицательного мутанта NSF (NSF E329Q) приводила к ингибированию аутофагического потока (фиг. S1 C и D ). Аутофагическая активность клеток, экспрессирующих GFP-LC3, также отслеживалась с помощью анализа ImageStream в качестве альтернативного подхода для определения влияния на аутофагию при нокдауне αSNAP (18).Измеряя интенсивность ярких деталей (BDI), которая сочетает в себе особенности текстуры и интенсивности (19), мы показываем, что аутофагический поток ингибируется при нокдауне αSNAP (Рис. 1 C и Рис. S1 G ). Чтобы исключить возможность косвенного воздействия нокдауна αSNAP на аутофагию, клетки, лишенные αSNAP, трансфицировали siRNA-устойчивым αSNAP, слитым с его N-концом с помощью тега FLAG. Эта обработка привела к полному восстановлению аутофагии в клетках, экспрессирующих siRNA-резистентный FLAG-αSNAP (полная линия на рис.1 D ) по сравнению с восстановлением соседних нетрансфицированных клеток (пунктирная линия на фиг. 1 D ). Восстановление аутофагического потока с помощью siRNA-устойчивого FLAG-αSNAP было также очевидно, когда клеточные лизаты подвергались Вестерн-блоттингу (рис. 1 E ).
Рис. 1. ПраймингSNARE необходим для аутофагического потока. ( A , Left ) Клетки HeLa трансфицировали ненаправленной (контрольной) siRNA (не tgt) или αSNAP siRNA (siαSNAP) с использованием реагента для трансфекции DharmaFECT 1.Через 48 часов клетки инкубировали в течение 4 часов в отсутствие или в присутствии 0,1 мкМ BafA, лизировали экстракционным буфером RIPA и затем анализировали вестерн-блоттингом. ( A , Right ) По крайней мере четыре независимых испытания были оценены количественно с использованием программного обеспечения Image Studio Lite, нормализованного по контрольному образцу без нацеливания. Значения представляют собой средние значения ± SE. ( B , Left ) Клетки HeLa обрабатывали, как в A , фиксировали 100% метанолом, иммуноокрашивали антителами против LC3 и p62 и анализировали с помощью конфокального микроскопа Olympus.(Масштабная шкала, 20 мкм.) Большое увеличение окрашенных клеток представлено в колонке Right . ( B , Right ) По меньшей мере 80 контрольных клеток (не обработанных BafA) были количественно определены с использованием программного обеспечения Imaris × 64 8.2.0, Bitplane. *** P <0,0001. Значения представляют собой средние значения ± SE. ( C ) Клетки HeLa, стабильно экспрессирующие GFP-LC3, обрабатывали, как в A , затем собирали трипсином, фиксировали 100% метанолом и окрашивали DAPI. Затем клетки подвергали анализу ImageStream, как описано в Materials and Methods .( Left ) Четыре независимых испытания были проанализированы и количественно оценены с помощью программного обеспечения IDEAS v. 6.2. Значения представляют собой средние значения ± SE. ( справа ) Типичная гистограмма BDI анализа ImageStream. ( D ) Клетки HeLa трансфицировали, как в A , и через 24 часа трансфецировали siRNA-устойчивым FLAG-меченным αSNAP с использованием реагента для трансфекции jetPEI в течение следующих 24 часов. Затем клетки инкубировали в течение 4 ч в отсутствие или в присутствии 0,1 мкМ BafA, фиксировали 100% метанолом, окрашивали антителами против p62 и против FLAG и анализировали с помощью конфокального микроскопа Olympus.(Шкала 20 мкм.) ( E ) Клетки обрабатывали, как в D , лизировали экстракционным буфером RIPA и затем анализировали вестерн-блоттингом.
Формирование аутофагосом не требует прайминга SNARE.
Чтобы определить, являются ли связанные с аутофагией структуры, накопленные в ответ на нокдаун αSNAP, открытыми фагофорами или запечатанными аутофагосомами, мы выделили мембраны из клеток, экспрессирующих GFP-LC3, и подвергли их обработке протеиназой K в присутствии или в отсутствие детергента Triton Х-100 (рис.2 A , Левая колонка ). Можно ожидать, что GFP-LC3 будет защищен от обработки протеазой только в том случае, если он находится в закрытых аутофагосомах. В этих экспериментах использовался Тритон Х-100 для растворения мембран, подвергая защищенные белки действию протеазы. Примечательно, что индуцированное протеазой расщепление экспонированного GFP-LC3, как известно, приводит к деградации LC3 и накоплению свободных молекул GFP (20). Как показано на фиг. 2 A ( правый столбец ), нокдаун αSNAP приводил к накоплению защищенных протеазой LC3 и p62, что свидетельствует о накоплении запечатанных аутофагосом.В клетках, трансфицированных нецелевой siRNA, защищенные LC3 и p62 выявлялись только в относительно небольших количествах, что согласуется с представлением о том, что в стационарных условиях количество присутствующих аутофагосом невелико. В качестве контроля для этой системы клетки инкубировали в присутствии BafA (который, как известно, накапливает зрелые аутофагосомы и автолизосомы), в результате чего накапливались защищенные LC3II и p62 как в контрольных, так и в αSNAP-нокдаун клетках (рис. 2 A ). Взятые вместе, эти результаты подтверждают, что прайминг SNARE важен в основном для поздних стадий аутофагии, но остается незаменимым для образования двухмембранной аутофагосомы.
Рис. 2.Формирование аутофагосом не требует прайминга SNARE. ( A , слева ) Изображение схемы анализа защиты, описанной в «Материалы и методы» . ( A , Right ) Клетки HeLa, стабильно экспрессирующие GFP-LC3, трансфицировали ненаправленной (контрольной) siRNA (не tgt) или αSNAP siRNA (siαSNAP) с использованием реагента для трансфекции DharmaFECT 1. Через 48 часов клетки инкубировали в течение 4 часов в отсутствие или в присутствии 0,1 мкМ BafA, а затем гомогенизировали, центрифугировали и подвергали обработке протеиназой K и тритоном X-100, как описано в Materials and Methods .Уровни белка определяли количественно с помощью программного обеспечения Image Studio Lite. ( B , Top иммунофлуоресцентный анализ) Клетки HeLa трансфицировали ненаправленной (контрольной) siRNA (не tgt), siRNA αSNAP (siαSNAP), siRNA трех членов семейства GABARAP (siGABARAPs) или с помощью siαSNARAP и обоих siαSNARAP. Реагент для трансфекции DharmaFECT 1. Через 48 ч клетки фиксировали 100% метанолом, иммуноокрашивали антителами против LC3 и против WIPI2 и анализировали с помощью конфокального микроскопа Olympus.(Масштабная шкала, 20 мкм.) Большое увеличение окрашивания WIPI представлено в правой колонке . ( B , Нижняя схема ) Изображение связанных с аутофагией тел, обнаруженных при различных обработках. ( C , Top изображения) Клетки HeLa пересекали, как в A . Через 48 ч клетки фиксировали 100% метанолом, иммуноокрашивали антителами против LC3 и p62 и подвергали двухцветной визуализации 3D STORM, как описано в Материалы и методы .( C , Нижний график ) Количественная оценка соотношения морфологий LC3- и p62-точек; n = 25. ( D , слева ) Клетки HeLa, стабильно экспрессирующие GFP-LC3, трансфицировали, как в A . Криосрезы фиксированных клеток иммуно метили антителами против GFP и анализировали с помощью ТЕМ, как описано в Materials and Methods . ( D , справа ) Анализ структур, меченных GFP, с помощью программного обеспечения ImageJ; n > 40, *** P <0.004. Значения представляют собой средние значения ± SE.
Дополнительное доказательство возможности того, что везикулы, которые накапливаются при истощении αSNAP, являются зрелыми аутофагосомами, было получено путем мониторинга WIPI2 (раннего фагофорного маркера, который отделяется от фагофора при образовании аутофагосомы) (21). Как показано на фиг. 2 B и фиг. S2 A , нокдаун всех членов семейства GABARAP ведет к большому накоплению меченных LC3 везикул, которые содержат WIPI2 (22). Напротив, везикулы, меченные LC3, которые накапливались в ответ на нокдаун αSNAP, были только частично помечены WIPI2, что указывает на то, что большинство везикул, которые накапливаются в этих условиях, представляют собой более позднюю стадию образования аутофагосом (рис.2 B и рис. S2 A ). Более того, нокдаун αSNAP вместе с GABARAP ведет к накоплению позитивной структуры LC3 и WIPI2, подтверждая представление о том, что αSNAP действует после образования аутофагосом (Fig. 2 B и Fig. S2 A ).
Затем точки, меченные LC3 и p62, были проанализированы с помощью микроскопии сверхвысокого разрешения. Пункты p62, которые были полностью покрыты LC3, что указывает на запечатанные аутофагосомы, были более распространены после нокдауна αSNAP по сравнению со структурами, в которых экспонировался сигнал p62 (рис.2C и фильмы S1 и S2). Как и ожидалось, в контрольных ячейках были изображены равные популяции двух структурных типов.
Для дальнейшего определения природы мембранных структур, которые накапливаются при истощении αSNAP, клетки, стабильно экспрессирующие GFP-LC3, анализировали с помощью корреляционной световой иммуноэлектронной микроскопии (фиг. 2 D и фиг. S2 B ). Очевидно, нокдаун αSNAP приводил к накоплению мембранных везикул, меченных GFP-LC3. LC3 был обнаружен как на внутренней (просвет), так и на внешней (цитозольной) сторонах этих мембран, указывая тем самым, что эти везикулы действительно являются запечатанными аутофагосомами с двойной мембраной.В отличие от контрольных клеток, в клетках, разрушенных для αSNAP, обнаруженные нами мембранные структуры, меченные GFP-LC3, были закрыты, и не было обнаружено значительных различий в средних размерах аутофагосом между контрольными клетками и клетками, выключенными αSNAP (рис. S2 B и С ). Более того, более подробная клеточная морфология, полученная с помощью просвечивающей электронной микроскопии, показала, что нокдаун αSNAP приводит к накоплению двойных мембранных аутофагосом, поглощающих цитоплазматический материал (рис.S2 C ).
αSNAP необходим для прайминга связанных с аутофагией комплексов SNARE.
Различные подходы были использованы для анализа способности зрелых аутофагосом, которые накапливаются при истощении αSNAP, сливаться с лизосомами. Во-первых, мы использовали иммунофлуоресцентный анализ для мониторинга, в отсутствие αSNAP, накопления аутофагосом, меченных LC3 (рис. 3 A ) или GFP-GATE16 (рис. S1 A ), которые были совместно локализованы с анти– Лизосомы, меченные LAMP1.Эти аутофагосомы действительно были обнаружены только в непосредственной близости от лизосомы, что указывает на нарушение слияния мембран между этими органеллами. Примечательно, что siRNA αSNAP не влияла на закисление лизосом, исключая возможность косвенного воздействия на активность лизосом (рис. S3 A ).
Рис. 3.αSNAP необходим для праймирования связанных с аутофагией комплексов SNARE. ( A , Left ) Клетки HeLa трансфицировали ненаправленной (контрольной) siRNA (не tgt) или αSNAP siRNA (siαSNAP) с использованием реагента для трансфекции DharmaFECT 1.Через 48 ч клетки фиксировали 100% метанолом, иммуноокрашивали антителами против LC3 и против LAMP1, а затем анализировали с помощью конфокального микроскопа Olympus. (Масштабная шкала, 20 мкм.) Большое увеличение окрашенных клеток представлено в колонке Right . ( A , справа ) Профили интенсивности отмеченных линий в столбце с большим увеличением были получены с помощью аналитического программного обеспечения Olympus FluoView FV 1000. ( B ) Клетки HeLa трансфицировали и обрабатывали, как в A , лизировали экстракционным буфером RIPA, нагревали до указанных температур и анализировали вестерн-блоттингом.nt , ненаправляющая (контрольная) миРНК. ( C ) Клетки HeLa трансфицировали, как в A , обрабатывали NEM и DTT или без них, лизировали буфером для лизиса SNARE и иммунопреципитировали антителами Vamp8. ( Левый ) Схема иммунопреципитации, как подробно описано в Материалы и методы . ( Right Top ) IP с антителами Vamp8. ( Нижний ) TCL, общие клеточные лизаты; Ат — антитела без инкубации лизата; ИП, иммунопреципитация. Более подробная информация представлена в «Материалы и методы» .
Затем мы исследовали образование комплексов SNARE, которые обеспечивают слияние мембран между аутофагосомами и лизосомами. Для этого мы воспользовались тем фактом, что комплексы SNARE часто устойчивы к SDS (23). Как показано на фиг. 3 B , когда образцы, полученные из клеток, обработанных siRNA αSNAP, разделяли на SDS / PAGE, было обнаружено, что синтаксин 17 мигрирует в виде большого устойчивого к SDS комплекса, который диссоциирует при кипячении. Затем с помощью метода иммунопреципитации мы исследовали эффект нокдауна αSNAP на связанный с аутофагией комплекс SNARE, состоящий из синтаксина 17, SNAP29 и VAMP8 (12).Используя антитела против VAMP8, мы обнаружили, что этот комплекс SNARE иммунопреципитируется только при нокдауне αSNAP или когда общий алкиллятор N -этилмалеимид (NEM) был использован для блокирования активности NSF (24) (рис. 3 C ). Когда мы использовали антитела против синтаксина 17 для иммунопреципитации комплекса, коиммунопреципитировался только SNAP29 (рис. S3 B ). Важно отметить, что накопление комплекса Vamp4 – синтаксин6 – синтаксин16 – Vti1, участвующего в эндоцитарном пути, также было очевидно при нокдауне αSNAP (рис.S3 C ). Кроме того, другие пути внутриклеточного транспорта, такие как лиганд-индуцированный эндоцитоз EGFR, а также перемещение ManII из эндоплазматического ретикулума (ER) в Гольджи, ингибировались при нокдауне αSNAP, что указывает на общий эффект на процессы слияния мембран в этих условиях (рис. S4). ). Взятые вместе, эти результаты показали, что αSNAP преимущественно необходим для праймирования комплекса SNARE, необходимого для слияния аутофагосомной и лизосомной мембран, тогда как образование аутофагосом менее чувствительно к его отсутствию.
Транслокация Syntaxin17 и Atg9 в аутофагосому не зависит от SNARE-прайминга.
Syntaxin17, как сообщается, перемещается из митохондрий в зрелые аутофагосомы (25). Поэтому мы исследовали, зависит ли такая транслокация от αSNAP. Анализ конфокальной микроскопии показал, что нокдаун αSNAP не влияет на транслокацию синтаксина 17 в аутофагосомы (Fig. 4 A ). Субклеточное фракционирование методом флотации в градиенте сахарозы показало, что в контрольных клетках синтаксин 17 кофракционируется в основном с митохондриальным маркером VDAC3 (рис.4 В , фракция 6). Однако небольшое количество софракционируется с аутофагосомными маркерами LC3 и p62, что соответствует небольшому количеству аутофагосом, присутствующих в этих условиях (фиг. 4 B , фракции 3 и 4). Поскольку нокдаун αSNAP ведет к накоплению аутофагосом, мы наблюдали — как и ожидалось — заметное увеличение синтаксина 17, кофракционированного с аутофагосомными маркерами (Fig. 4 B ), предполагая, что его транслокация в аутофагосомы не зависит от αSNAP.Примечательно, что нокдаун αSNAP не влиял на миграцию VAMP8 и SNAP29 по градиенту сахарозы. В целом, эти находки подтверждают, что рекрутирование syntaxin17 на мембрану аутофагосом не зависит от αSNAP.
Рис. 4.Транслокация syntaxin17 и Atg9 в аутофагосому не зависит от SNARE-прайминга. ( A , Left ) Клетки HeLa трансфицировали ненаправленной (контрольной) siRNA (не tgt) или αSNAP siRNA (siαSNAP) с использованием реагента для трансфекции DharmaFect 1.Через 24 часа клетки трансфицировали синтаксином 17, меченным FLAG, с использованием реагента для трансфекции jetPEI в течение дополнительных 24 часов. Затем клетки фиксировали 100% метанолом, окрашивали антителами против LC3 и против FLAG и анализировали с помощью конфокального микроскопа Olympus. (Масштабная шкала, 20 мкм.) Правая колонка показывает окрашенные клетки с большим увеличением. ( Правый ) По крайней мере, три независимых испытания были количественно оценены с использованием программного обеспечения Imaris × 64 8.2.0, Bitplane. NS, не имеет значения. Значения представляют собой средние значения ± SE.( B , вверху слева ) Клетки HeLa трансфицировали, как в A . Через 48 ч клетки гомогенизировали, помещали в градиент сахарозы, как описано в Materials and Methods , и количественно определяли с помощью программного обеспечения Image Studio Lite. ( Вверху справа ) Схема анализа флотации, как подробно описано в Материалы и методы . ( Нижний ) Уровни белка во фракциях определяли количественно с использованием программного обеспечения Image Studio Lite. ( C ) Клетки HeLa трансфицировали, как в A , фиксировали 4% параформальдегидом, иммуноокрашивали антителами против Atg9, анти-p115 и анти-p62 и анализировали с помощью конфокального микроскопа Olympus.(Масштабная полоса, 20 мкм.) Большое увеличение окрашенных клеток представлено в колонке справа каждой панели.
Atg9, единственный идентифицированный трансмембранный белок, необходимый для образования полных аутофагосом, синтезируется в ER и перемещается между Гольджи и эндосомами (26, 27). Мы использовали субклеточное фракционирование и иммунофлуоресцентную микроскопию, чтобы изучить влияние нокдауна αSNAP на субклеточную локализацию Atg9. В контрольных условиях Atg9 кофракционировал с ERGIC-53, маркером промежуточного компартмента ER – Golgi (рис.4 B , фракция 5), а также с аутофагосомными маркерами p62 и LC3 (рис. 4 B , фракции 3 и 4). Аналогичные результаты были получены с помощью анализа конфокальной микроскопии (рис. 4 C , правая колонка ). После нокдауна αSNAP способность Atg9 достигать аутофагосом сохранялась (рис. 4 B ), хотя он был диспергирован вместе с маркером Гольджи p115, периферическим ассоциированным с Гольджи белком, внутри клетки, вероятно, из-за дисперсии Гольджи на Ингибирование αSNAP (рис.4 C , Левая колонка ). Эти результаты подтверждают, что вклад Atg9 в биогенез аутофагосом не зависит от примирования SNARE. Это открытие согласуется с сообщением, показывающим, что пузырьки Atg9 обнаруживаются вблизи аутофагических мембран, но не сливаются с ними (26).
Низкий уровень αSNAP достаточен для частичного стимулирования аутофагии в условиях голода.
Затем мы исследовали влияние истощения αSNAP на аутофагию, вызванную голоданием.Как показано на фиг. 5, нокдаун αSNAP приводил к ингибированию аутофагического потока также в голодных клетках (фиг. 5 A и B и фиг. S1 C — F ). По-видимому, эффект истощения αSNAP на аутофагию, вызванную голоданием, был ниже, чем эффект, наблюдаемый в контрольных условиях. Статистический анализ показал значительное увеличение количества аутофагосом, но не размера в клетках после трансфекции siRNA αSNAP (фиг. 5 B и фиг. S2 C и S5 A ).Чтобы определить, достаточны ли оставшиеся молекулы αSNAP для нацеливания NSF на мембраны, обеспечивающие праймирование SNARE при голодании, мы разделили клетки, фракционируя их на высокоскоростной супернатант (HSS) (цитозольная фракция) и высокоскоростной осадок (HSP) (мембранная фракция). . Мы обнаружили, что и αSNAP, и NSF накапливаются в осадках клеток, не трансфицированных миРНК, и что при голодании уровни αSNAP и NSF, обнаруженные во фракции осадка, выше, чем в условиях кормления (рис.5 С ). Однако при истощении αSNAP, тогда как NSF был обнаружен как в супернатанте, так и в осадке, остаточные количества αSNAP были обнаружены только в осадке. Это открытие согласуется с ролью αSNAP как адапторного белка, ограничивающего способность NSF связывать мембраны. Кроме того, при голодании уровни синтаксина 17 и SNAP29 молекул SNARE в осадке в клетках с нокдауном αSNAP снижались, указывая на разборку комплекса SNARE (рис. 5 C ).Более того, как в отсутствие, так и в присутствии NEM количество SNAP29, коиммунопреципитированного с синтаксином 17, было ниже в голодающих клетках с истощением αSNAP, чем в клетках в условиях питания (фиг. S5 B ). Таким образом, наши результаты свидетельствуют о том, что при голодании низкие уровни αSNAP способны связывать молекулы NSF, чтобы частично способствовать праймингу SNARE, необходимому для аутофагии.
Рис. 5.Низкий уровень αSNAP достаточен для частичного стимулирования аутофагии в условиях голодания.( A , Left ) Клетки HeLa трансфицировали ненаправленной (контрольной) siRNA (не tgt) или siRNA αSNAP с использованием реагента для трансфекции DharmaFECT 1. Через 48 часов клетки инкубировали в течение 4 часов в нормальной ростовой среде или EBSS в отсутствие или в присутствии 0,1 мкМ BafA, лизировали экстракционным буфером RIPA и затем анализировали вестерн-блоттингом. ( A , Right ) По крайней мере четыре независимых испытания были оценены количественно с использованием программного обеспечения Image Studio Lite, нормализованного по контрольному образцу без нацеливания.Значения представляют собой средние значения ± SE. Голод, голодные условия. ( B , Left ) Клетки HeLa обрабатывали, как в A , фиксировали 100% метанолом, иммуноокрашивали антителами против LC3, анализировали конфокальным микроскопом Leica с использованием одного и того же порога для всех образцов и деконволюционировали с помощью программного обеспечения Huygens. . Проекция Z-стека была сделана с помощью программного обеспечения ImageJ. (Масштабная шкала, 10 мкм.) Большое увеличение окрашенных клеток представлено на справа . ( Средний ) Количественный анализ точек LC3 с использованием программного обеспечения LAS X, Leica Microsystems. n > 1000, *** P <0,0001. Значения представляют собой средние значения ± SE. ( Right ) Клетки HeLa, стабильно экспрессирующие GFP-LC3, обрабатывали, как в A , затем собирали трипсином, фиксировали 100% метанолом и окрашивали DAPI. Затем клетки подвергали анализу ImageStream, как описано в Materials and Methods . Четыре независимых испытания были проанализированы и количественно оценены с помощью программного обеспечения IDEAS v. 6.2. ( C ) Клетки HeLa трансфицировали, как в A , гомогенизировали и фракционировали, как описано в «Материалы и методы» .Общие гомогенаты клеток представлены на Справа .
Обсуждение
Мало что известно об участии прайминга SNARE в аутофагическом процессе и, в частности, в биогенезе аутофагосом. В этом исследовании, подавляя αSNAP — белок, который служит важным адаптером почти для всех процессов, включающих слияние внутриклеточных мембран — мы показали, что хотя аутофагосомы все еще образуются в его отсутствие, их способность слиться с лизосомами нарушена.Это привело к ингибированию аутофагического потока, проявляющегося в накоплении запечатанных аутофагосом, маркированных LC3, p62 и синтаксином 17. Мы также продемонстрировали, что αSNAP не нужен для ассоциации синтаксина 17 и Atg9 с аутофагосомной мембраной. Взятые вместе, наши результаты подразумевают, что разборка SNARE важна для слияния аутофагосомы с лизосомой, но не для инициации фагофоров или снабжения мембран во время биогенеза аутофагосом. Примечательно, однако, что наши результаты не исключают необходимости SNARE-зависимого слияния мембран для начальных этапов биогенеза аутофагосом.
Прайминг SNARE управляется NSF, ферментом, для которого αSNAP действует как структурный адаптер. Обе активности необходимы для выживания клеток. Здесь мы использовали подход siRNA для изучения роли прайминга SNARE в аутофагии и решили сосредоточиться на адапторной молекуле αSNAP, поскольку нокдаун фермента NSF привел к более слабому влиянию на аутофагический поток, скорее всего, из-за остаточной ферментативной активности NSF. Важно отметить, что нокдаун αSNAP одинаково и беспристрастно влияет не только на конкретную молекулу SNARE, но и на все события прайминга SNARE, предотвращая, таким образом, несбалансированные изменения в других процессах внутриклеточного трафика.Неожиданно наши результаты показали, что, хотя нокдаун αSNAP подавляет аутофагический поток, он не ингибирует образование аутофагосом, подтверждая представление о том, что биогенез аутофагосом менее чувствителен к потере прайминга SNARE.
Syntaxin17 недавно был идентифицирован как важная молекула SNARE для аутофагии. Первоначально сообщалось, что он включается в зрелые аутофагосомы, чтобы способствовать слиянию аутофагосомной и лизосомальной мембран (12, 13), тогда как более недавнее исследование показало, что он может принимать участие в формировании изолирующей мембраны (28).Интересно, что синтаксин 17 также был обнаружен в митохондриях, где он способствует делению митохондрий независимо от своего домена SNARE, а также гомеостазу кальция в ER и переходит к своей аутофагической роли при голодании (25). Наши настоящие результаты показали, что синтаксин 17 находится в митохондриях в контрольных условиях, но в отсутствие αSNAP он накапливается на аутофагосомах, указывая на то, что его доставка к этим органеллам не требует примирования SNARE. Следовательно, кажется, что его накопление на аутофагосомах просто отражает их неспособность созревать и сливаться с лизосомами.Примечательно, что мы показываем, что нокдаун αSNAP в нашей системе сопровождается накоплением других, не связанных с аутофагией, комплексов SNARE, а также ингибированием общих процессов трафика, что указывает на то, что были затронуты множественные SNARE-зависимые процессы трафика.
Участие Atg9 в биогенезе аутофагосом широко изучалось как в дрожжах, так и в клетках млекопитающих (26, 27). У млекопитающих он локализуется в кратковременной близости к фагофорам, но не встраивается в них (26) и рециркулируется в эндосомные компартменты из Гольджи, где, по-видимому, наиболее обильно (29).Наши данные показали, что вклад Atg9 в биогенез аутофагосом не зависит от прайминга SNARE. В то время как локализация Atg9 в клетке была затронута при ингибировании прайминга SNARE (вероятно, из-за дисперсии Гольджи), его локализация в аутофагосомах осталась неизменной, что согласуется с представлением о том, что связанные с Atg9 везикулы не сливаются с аутофагосомами (26, 30) .
Голодание — это хорошо зарекомендовавший себя индуктор аутофагии, который, как известно, ускоряет биогенез аутофагосом и их последующую лизосомную деградацию.Тем не менее мы обнаружили, что нокдаун αSNAP в голодных клетках приводит к частичному ингибированию аутофагического потока по сравнению с клетками, выращенными в полной среде. Пытаясь предоставить доказательства для объяснения этого феномена, мы предоставляем данные, предполагающие, что при голодании более высокие уровни NSF нацелены на мембраны, где они могут в достаточной степени способствовать праймингу SNARE, необходимому для аутофагии. Таким образом, мы предполагаем, что при голодании αSNAP и NSF нацелены на комплексы SNARE, специфически участвующие в слиянии аутофагосомно-лизосомальной мембраны.Механизм, отвечающий за регулирование этого процесса, будет предметом будущих исследований.
Взятые вместе, наши результаты показали, что слияние мембран аутофагосом с лизосомой чувствительно к ингибированию прайминга SNARE, тогда как образование аутофагосом устойчиво к его потере. Вероятный сценарий состоит в том, что низкие уровни NSF и αSNAP позволяют праймировать комплексы SNARE, необходимые для образования аутофагосом, но не SNAREs, которые действуют в слиянии аутофагосома-лизосома; с другой стороны, наши данные могут указывать на то, что процесс в достаточной степени поддерживается зарождающимися неспаренными молекулами SNARE.
Материалы и методы
Дополнительные методы см. В дополнительной информации .
Культура клеток и трансфекция.
Клетки HeLa выращивали на среде αMEM с добавлением 10% FCS при 37 ° C в 5% CO. 2 . Для подавления siRNA субконфлюентные клетки HeLa трансфицировали различными пулами siRNA или олигонуклеотидами с использованием реагента для трансфекции DharmaFect 1 (Dharmacon) в соответствии с инструкциями производителя. Эксперименты проводили через 48 ч после трансфекции.Трансфекцию плазмид для сверхэкспрессии белка проводили с использованием реагента для трансфекции jetPEI (трансфекция Polyplus) в соответствии с инструкциями производителя. Условия голодания достигаются путем трехкратной промывки клеток PBS и их инкубации в среде сбалансированного солевого раствора Эрла (EBSS) при 37 ° C в течение 4 часов. Лизосомную деградацию ингибировали инкубацией клеток в присутствии 100 нМ BafA (LC Laboratories). Полные экстракты клеток получали с использованием буфера для экстракции RIPA (0,1 М NaCl, 5 мМ ЭДТА, 0.1 M Na 2 HPO 4 / NaH 2 PO 4 pH 7,5, 1% тритон, 0,5% DOC, 0,1% SDS) со смесью ингибиторов протеаз (Calbiochem). Уровни белка измеряли с помощью программного обеспечения Image Studio Lite. Уровни деградации белка рассчитывали путем деления уровней белка в образцах, обработанных BafA, на уровни в необработанных образцах.
Флуоресцентная микроскопия.
Клетки высевали на стерильные покровные стекла (диаметром 13 мм) и культивировали в указанных условиях.Клетки фиксировали 4% параформальдегидом в PBS в течение 10 минут и повышали проницаемость с помощью 0,1% Triton X-100 в течение 8 минут. При использовании антител против LC3 клетки фиксировали и пермеабилизировали холодным метанолом в течение 7 минут при -20 ° C. Клетки блокировали инкубацией с 10% FCS в PBS в течение 30 минут при комнатной температуре с последующей инкубацией в течение 1 часа с первичным антителом. Затем клетки инкубировали со вторичным антителом в течение 30 минут. Конфокальные изображения были получены с помощью лазерно-сканирующего конфокального микроскопа FV1000 Olympus, оснащенного UPLS APO 60 × N.A. Масляная иммерсионная линза 1.35 или конфокальный лазерный сканирующий микроскоп Leica TCS SP8, оснащенный масляной иммерсионной линзой HCPL APO CS2 63 × / 1.40, как показано. Изображения анализировали с помощью программного обеспечения FluoView FV1000 (Olympus) или программ Huygens и LASX, соответственно. Порог количественного определения аутофагосом во всех образцах составлял 0,031 мкм 3 . Визуализацию живых клеток получали с помощью микроскопа DeltaVision (Applied Precision). Значения колокализации между различными маркерами были проанализированы по крайней мере в двух отдельных экспериментах с помощью Imaris × 64 8.2.0 программное обеспечение, Bitplane.
Благодарности
Благодарим Владимира Кисса за помощь в конфокальной микроскопии. Это исследование было частично поддержано Израильским научным фондом (грант 1247/15), Фондом наследия наследия (грант 1935/16) и Фондом Минервы при финансовой поддержке Федерального министерства образования и исследований Германии.
Сноски
Авторы: A.A. и З. спланированное исследование; A.A., S.L.-Z., Z.P. и T.D. проводили исследования; А.А., Т.Д. и З.Е. проанализированные данные; и А.А. и З. написал газету.
Авторы заявляют об отсутствии конфликта интересов.
Эта статья представляет собой прямое представление PNAS. СИДЕЛ. является приглашенным редактором по приглашению редакционной коллегии.
Эта статья содержит вспомогательную информацию на сайте www.pnas.org/lookup/suppl/doi:10.1073/pnas.1705572114/-/DCSupplemental.
- Copyright © 2017 Автор (ы). Опубликовано PNAS.
Недостаточное подкисление аутофагосом способствует выживанию и росту стрептококков группы А в эндотелиальных клетках
РЕЗЮМЕ
Стрептококк группы А (ГАЗ) является важным патогеном человека, и его проникновение через кровеносные сосуды критически важно при серьезных событиях, таких как бактериемия полиорганная недостаточность. Хотя GAS был идентифицирован как внеклеточная бактерия, интернализация GAS в нефагоцитарные клетки может обеспечить стратегию ухода от иммунного надзора и уничтожения антибиотиками.Однако сообщалось также, что GAS вызывает аутофагию и эффективно убивается в слитых с лизосомами аутофагосомах в эпителиальных клетках. В этом исследовании мы показываем, что GAS может реплицироваться в эндотелиальных клетках и что стрептолизин O необходим для роста GAS. Бактериальную репликацию можно подавить путем изменения экспрессии гена GAS в кислой среде перед интернализацией в эндотелиальные клетки. Ингибирующий эффект на репликацию GAS может быть отменен обработкой бафиломицином A1, специфическим ингибитором вакуолярного типа H + -АТФазы.По сравнению с эпителиальными клетками, в которых закисление вызывает опосредованный аутофагией клиренс GAS, в эндотелиальных клетках наблюдался дефект закисления GAS-содержащих пузырьков. Следовательно, эндотелиальные клетки не в состоянии поддерживать низкий уровень pH в GAS-содержащих аутофагосомах, что позволяет репликации GAS внутри LAMP-1- и LC3-положительных везикул. Кроме того, обработка эпителиальных клеток бафиломицином A1 приводила к нарушению клиренса GAS за счет аутофагии с последующим внутриклеточным ростом бактерий.Следовательно, низкий pH является ключевым фактором опосредованного аутофагией подавления роста GAS внутри эпителиальных клеток, в то время как дефектное закисление GAS-содержащих пузырьков приводит к росту бактерий в эндотелиальных клетках.
ВАЖНОСТЬ Предыдущие сообщения показали, что ГАЗ может вызывать аутофагию и эффективно убивается в слитых с лизосомами аутофагосомах в эпителиальных клетках. В эндотелиальных клетках, напротив, индукции аутофагии недостаточно для уничтожения ГАЗ. В этом исследовании мы представляем первое доказательство того, что низкий pH необходим для предотвращения внутриклеточного роста GAS в эпителиальных клетках и что этот механизм неисправен в эндотелиальных клетках.Обработка GAS низким pH изменяла скорость роста GAS и экспрессию генов факторов вирулентности и приводила к повышенной чувствительности GAS к внутриклеточному уничтожению лизосом. Наши результаты показывают существование различных механизмов защиты хозяина от инвазии GAS между эпителиальными и эндотелиальными клетками.
ВВЕДЕНИЕ
Стрептококк группы А (ГАЗ) (Streptococcus pyogenes) является обычным патогеном человека и вызывает широкий спектр человеческих заболеваний. Несмотря на доступность эффективных противомикробных препаратов, в последние годы во всем мире наблюдается рост случаев серьезной инвазивной инфекции ГАЗ (1–5).Хотя GAS считается внеклеточным патогеном, интернализация GAS неиммунными клетками может дать бактериям возможность избежать фагоцитоза и уничтожения антибиотиками (6–9). Небольшое количество GAS может скрываться внутри клеток на стадии носительства у бессимптомных хозяев (10–13).
Аутофагия — это внутриклеточный деградационный процесс, который важен для уравновешивания источников энергии в критические периоды развития и в ответ на стресс, связанный с питанием. Аутофагосома устанавливается в месте взаимодействия эндоплазматического ретикулума и митохондрий (14) и образована удлиненной двойной мембранной структурой, называемой изолирующей мембраной, которая окружает груз, а затем закрывается, образуя большую вакуольоподобную структуру.Долгоживущие белки или поврежденные органеллы, такие как эндосомы, лизосомы или митохондрии, становятся мишенью изолирующей мембраны и разрушаются в образующихся аутофаголизосомах (15–18). Помимо внутриклеточных компонентов, аутофагия также важна для защиты от вторжения патогенов (19–22), хотя она не всегда полезна для клеток-хозяев (23, 24). Подобно эндосомам и фагосомам, аутофагосомы также полагаются на функцию лизосом для разложения груза. Лизосома содержит пищеварительные гидролитические ферменты и микроокружение с низким pH.Эта среда поддерживается протонными насосами и каналами хлорид-ионов и необходима для активности ферментов (25). Однако, помимо необходимости для функции фермента, эта кислотность сама по себе может быстро подавлять рост патогенов в поздних эндосомах / фагосомах при слиянии с лизосомами (26).
Ранее было показано, что после проникновения в эпителиальные клетки GAS выходит из эндосомы в цитоплазму и захватывается в компартментах, подобных аутофагосомам, которые, в свою очередь, сливаются с лизосомами, что приводит к гибели GAS (20, 22, 27–29). ).Тем не менее глобально распространенный серотип M1T1 GAS может избегать аутофагии и эффективно реплицироваться в цитозоле инфицированных эпителиальных клеток (30). По сравнению с эпителиальными клетками, как M1, так и M3 GAS, как было обнаружено, проникают в эндотелиальные клетки (31–33). Помимо инвазии, M1 GAS также может выжить в эндотелиальных клетках (33). Однако причины того, почему и как GAS растет внутри эндотелиальных клеток, остаются неясными. В настоящем исследовании мы показываем, что GAS может реплицироваться в эндотелиальных клетках независимо от индукции аутофагии.Не только серотип M1, но и другие протестированные серотипы, включая M4, M6, M12 и M49, могут расти внутри эндотелиальных клеток. По сравнению с эпителиальными клетками, в которых подкисление вызывает опосредованный аутофагией клиренс GAS, низкий pH не поддерживается в GAS-содержащих аутофагосомах в эндотелиальных клетках. В этом исследовании мы показываем, что низкий pH важен для подавления выживания GAS и что снижение кислотности в эндотелиальных клетках ингибирует уничтожение лизосомных бактерий.
РЕЗУЛЬТАТЫ
Репликация GAS с различными серотипами М в эндотелиальных клетках.Предыдущее исследование показало клиренс ГАЗ путем аутофагии в эпителиальных клетках (22). Напротив, M1 GAS проникает в эндотелиальные клетки и растет в них (33). Однако механизм репликации GAS внутри эндотелиальных клеток остается неясным. В этом исследовании мы проверили судьбу GAS внутри эндотелиальных клеток. Мы инфицировали клетки HMEC-1 (линия 1 эндотелиальных клеток микрососудов человека) различными штаммами GAS в течение 1 часа и использовали гентамицин для уничтожения внеклеточных бактерий. Результаты показали, что внутриклеточное бактериальное количество штаммов GAS A20 (серотип M1), 125 (серотип M4), 712 (серотип M6), 733 (серотип M12) и NZ131 (серотип M49) показало зависящее от времени постинфекционное увеличение HMEC-1. клетки (рис.1А). В отличие от роста клеток HMEC-1, эти штаммы GAS не росли в эпителиальных клетках A549 (см. Фиг. S1 в дополнительном материале). Чтобы подтвердить, что частицы GAS существуют внутри клетки и не связаны с плазматической мембраной, клетки HMEC-1, инфицированные NZ131, окрашивали 4 ‘, 6’-диамидино-2-фенилиндолом (DAPI) и наблюдали под конфокальной микроскопией. Частицы GAS в цитоплазме, как обнаружено окрашиванием ДНК DAPI, увеличивались в зависимости от времени (рис. 1B). Репликация GAS внутри клеток HMEC-1 была дополнительно подтверждена с использованием поликлональных антител против GAS с последующим анализом проточной цитометрии (рис.1С и Г).
Рисунок S1
GAS не реплицируется в эпителиальных клетках. Клетки A549 были инфицированы серотипами M1 (штамм A20), M4, M6, M12 (клинически изолированные инвазивные штаммы 125, 712 и 733 [номера штаммов присвоены серийно] из серотипов M4, M6 и M12 соответственно) и M49 (NZ131 штамм) при MOI 5 в течение 1 часа. Внеклеточные бактерии были убиты обработкой гентамицином. Количество внутриклеточных бактерий определяли с помощью анализа образования колоний в различные моменты времени после заражения.Подсчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий через 2 часа после инфицирования. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. Скачать Рисунок S1, файл TIF, 0,2 МБ.Авторские права © 2015 Lu et al.Рис. 1.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Репликация GAS в эндотелиальных клетках. (A) Клетки HMEC-1 были инфицированы серотипом M1 (штамм A20), M4, M6, M12 (клинически изолированные инвазивные штаммы 125, 712 и 733 [номера штаммов присвоены серийно] серотипов M4, M6 и M12 соответственно) и M49 (штамм NZ131) при MOI 1 в течение 1 часа. Внеклеточные бактерии были убиты обработкой гентамицином. Количество внутриклеточных бактерий определяли с помощью анализа образования колоний в различные моменты времени после заражения.Подсчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий через 2 часа после инфицирования. Данные представляют собой средние значения ± стандартное отклонение (SD) (планки ошибок) из трех независимых экспериментов. (B) NZ131-инфицированные клетки HMEC-1 окрашивали DAPI и наблюдали с помощью конфокальной микроскопии через 1, 3 и 6 часов. Прутки, 10 мкм. (C) Клетки HMEC-1 инфицировали штаммом NZ131 при MOI 25 в течение 1 ч, а затем обрабатывали гентамицином. В различные моменты времени после инфицирования клетки окрашивали поликлональными антителами против GAS и анализировали проточной цитометрией.(C и D) Один набор гистограмм показан (C), и показаны средние значения плюс стандартное отклонение от трех независимых экспериментов (D). SSC, боковой разброс, представлен как степень детализации ячеек или внутренняя сложность.
Предыдущее сообщение показало, что стрептолизин O (SLO) необходим для повреждения эндосомальной мембраны и дополнительно способствует выходу ГАЗ из эндосомы в цитоплазму эпителиальной клетки (22). Кроме того, недавние сообщения показали, что SLO взаимодействует с NAD-гликогидролазой против уничтожения лизосом и продлевает внутриклеточное выживание GAS в кератиноцитах и макрофагах (34, 35).Чтобы определить, требуется ли SLO также для роста GAS в эндотелиальных клетках, мы исследовали бактериальный рост GAS дикого типа и с удаленным SLO. Результаты показали, что штамм A20 с удаленным SLO не может расти в эндотелиальных клетках (см. Фиг. S2A в дополнительном материале). Мы также протестировали штаммы JRS4 дикого типа (серотип M6) и мутантные штаммы с удаленным SLO, которые ранее были исследованы на эпителиальных клетках (22). Результаты показали, что JRS4 дикого типа рос в эндотелиальных клетках, тогда как мутантный штамм SLO не мог даже выжить в клетках (рис.S2B). Следовательно, SLO необходим для роста GAS в эндотелиальных клетках.
Рисунок S2
SLO необходим для роста GAS в эндотелиальных клетках. Клетки HMEC-1 инфицировали штаммом A20 или A20 с удаленным SLO (SW555) (A) и штаммом дикого типа JRS4 или JRS4 с удаленным SLO (B) при MOI, равном 1, в течение 1 часа. Внеклеточные бактерии были убиты обработкой гентамицином. Количество внутриклеточных бактерий определяли с помощью анализа образования колоний в различные моменты времени после заражения. Подсчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий через 2 часа после инфицирования.Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. Скачать Рисунок S2, файл TIF, 0,1 МБ.Авторские права © 2015 Lu et al.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Низкий pH влияет на рост GAS в эндотелиальных клетках.Потребление питательных веществ и накопление побочных продуктов метаболизма ощущается бактериями, которые изменяют экспрессию генов и фазу роста (36–38). Предыдущие сообщения показали, что pH был снижен в среде для долгосрочной культивирования GAS и что модифицированная среда с отрегулированным pH могла изменить экспрессию и выживаемость GAS-гена (39–43). Мы подтвердили изменение pH в бульонной среде во время роста, и результаты согласуются с предыдущими выводами о том, что pH постепенно снижается с ростом GAS с течением времени и что самый низкий уровень pH (pH ~ 5.5) был достигнут, когда рост бактерий достиг стационарной фазы (рис. 2А). Поэтому мы сравнили скорость роста эндотелиальных клеток между GAS в стационарной фазе и GAS, собранными в логарифмической фазе. Не было значительной разницы в эффективности интернализации ГАЗ между логарифмической и стационарной фазами (см. Рис. S3A в дополнительном материале). ГАЗ, собранный в стационарной фазе, рос внутри эндотелиальных клеток намного медленнее, чем ГАЗ, собранный в логарифмической фазе (рис. 2В). Чтобы исследовать, влияет ли низкий pH на рост GAS внутри эндотелиальных клеток, мы инфицировали клетки GAS, предварительно обработанным кислотой.Результаты показали, что без разницы в эффективности интернализации (фиг. S3B) предварительно обработанный кислотой GAS имел пониженную способность к росту и выживанию в эндотелиальных клетках (фиг. 2C).
Рисунок S3
Эффективность интернализации ГАЗА не зависит от pH культуральной среды. (A) Стационарная фаза и ранняя логарифмическая фаза GAS (штамм NZ131) были приготовлены, как описано в легенде к рис. 2. Бактерии затем подсчитывались с помощью анализа образования колоний и сравнивались с GAS, собранным перед уничтожением антибиотиком, для расчета эффективности интернализации. .Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. (B) После 1 ч инкубации свежей кислой или нейтральной среды TSBY, бактерии GAS собирали и добавляли к клеткам HMEC-1 на 30 мин. После трех промывок PBS добавляли гентамицин для уничтожения внеклеточных бактерий в течение дополнительных 30 минут и рассчитывали эффективность интернализации. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. Скачать Рисунок S3, файл TIF, 0,2 МБ.Авторские права © 2015 Lu et al.FIG 2Это статья в открытом доступе, распространяемая на условиях Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Непортированная лицензия, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Низкий pH влияет на рост GAS в эндотелиальных клетках и в бульоне TSBY. После ночного культивирования бактерии GAS (штамм NZ131) переносили в свежий бульон и культивировали при 37 ° C. (A) Бактериальный рост определяли при OD 600 в различные моменты времени. Супернатанты бактериальных культур собирали центрифугированием и фильтрацией в указанные моменты времени, и в супернатантах определяли pH.(B) Бактерии, культивируемые в течение ночи, использовали в качестве ГАЗ «стационарной фазы», а бактерии, обработанные свежей средой в течение 3 часов (обновленный ГАЗ), использовали как ГАЗ «ранней логарифмической фазы». Показанный здесь ГАЗ ранней логарифмической фазы использовался в качестве стандартной культуры для остальных экспериментов. (C) GAS-бактерии, обновленные в течение 3 часов, дополнительно инкубировали в свежем нейтральном бульоне или бульоне с низким pH в течение 1 часа, что обозначено как предварительная обработка кислотой. Клетки HMEC-1 инфицировали указанным GAS (B и C) при MOI 5 в течение 30 минут, а затем добавляли гентамицин для уничтожения внеклеточных бактерий.Бактерии собирали в указанные моменты времени после обработки гентамицином, и рассчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий (см. Фиг. S3 в дополнительном материале). (D) После культивирования в течение ночи бактерии GAS переносили в свежий или pH-отрегулированный бульон TSBY (pH ± 0,2). (E) Предварительно обработанные кислотой бактерии GAS были дополнительно перенесены в свежий нейтральный бульон. Рост бактерий в бульоне измеряли при OD 600 в различные моменты времени.(F) Клетки HMEC-1 предварительно обрабатывали бафиломицином A1 (Baf A) (100 нМ) в течение 1 часа и инфицировали указанным GAS в течение 30 минут. Затем проводили колониеобразующий анализ, как на панелях B и C. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. *, P <0,05; **, P <0,01.
Мы предположили, что изменение pH ощущается бактериями, которые затем замедляют рост, чтобы адаптироваться к изменениям окружающей среды. Чтобы подтвердить, что pH был причиной снижения роста бактерий, мы скорректировали уровни pH свежей бульонной среды и измерили рост GAS.Наши результаты показали, что кислая среда подавляет рост ГАЗ (рис. 2D). Когда GAS сначала инкубировали в нейтральной (pH 7) или кислой (pH 5,5) среде, а затем переносили в свежую нейтральную среду, результаты показали, что восстановление pH с pH 5,5 до нейтрального pH спасало скорость роста до уровней, наблюдаемых для необработанных кислотой ГАЗ (рис. 2Е). Кроме того, обработка эндотелиальных клеток бафиломицином A1 (Baf A), специфическим ингибитором вакуолярного типа H + -АТФазы, спасала рост бактерий, предварительно обработанных низким pH (рис.2F). Затем мы разделили GAS-инфицированные клетки на два типа: один мы назвали GAS-типом с низким ростом (L-тип; рис. 3Aa и b), а другой GAS-типом с высоким ростом (H-тип; рис. 3Ac и d). Результаты показали, что распределение предварительно обработанного кислотой GAS в эндотелиальных клетках ограничивалось паттерном L-типа, в то время как неподкисленный GAS демонстрировал распределение H-типа (фиг. 3Ae и B). Мы также подтвердили, что предварительно обработанный кислотой ГАЗ ограничивался LAMP-1-положительными вакуолями, поздним эндосомным или лизосомным маркером (рис. 3С).Эти данные показывают, что подавление роста GAS требует постоянного низкого pH в бактериальной культуральной среде, а также во внутриклеточной бактериальной вакуолярной среде.
FIG 3Низкое значение pH требуется для подавления репликации GAS в эндотелиальных клетках. (A) Внутриклеточный GAS (штамм NZ131), окрашенный DAPI, был разделен на две группы: с низким ростом (тип L) (а и b) и с высоким ростом (тип H) (с и d). После 6-часового заражения большинство инфицированных клеток HMEC-1 имеют структуру H-типа (e).Прутки, 10 мкм. (B) Клетки HMEC-1, инфицированные GAS (с предварительной обработкой кислотой или без нее), окрашивали DAPI, и процентное содержание клеток с инфекцией GAS и клеток с структурой H-типа рассчитывали с помощью конфокальной микроскопии. Наблюдалось более 100 клеток в каждом образце, и показаны средние значения ± стандартное отклонение из трех независимых экспериментов. **, P <0,01. (C) Клетки HMEC-1 инфицировали предварительно обработанным кислотой GAS, а затем фиксировали и окрашивали антителом против LAMP-1 и DAPI через 3 и 5 часов после инфицирования.Изображения наблюдали под конфокальной микроскопией. Прутки, 10 мкм.
Низкое значение pH изменяет экспрессию гена вирулентности GAS. Помимо воздействия на рост GAS, простая 1-часовая обработка кислотой может напрямую изменить экспрессию гена вирулентности GAS, в том числе spn ( nga ), slo , sagB и speB (рис. 4А). Гены slo, и sagB отвечают за производство гемолитических белков (SLO и SLS), которые повреждают структуру клеточной мембраны и помогают GAS ускользать из эндосом (22).Экспрессия гена spn ( nga ) продуцирует НАД-гликогидролазу, которая важна для вирулентности GAS (44) и для предотвращения убийства фаголизосом (34, 35). Наши результаты показали, что после обработки с низким pH GAS показал снижение внутриклеточной выживаемости. Без способности разрывать эндосомальную мембрану и противостоять антилизосомному перевариванию, предварительно обработанный кислотой GAS мог бы легко выводиться за счет эндосомальной деградации в эндотелиальных клетках.
FIG 4Низкий pH влияет на экспрессию гена фактора вирулентности GAS.(A) Обновленные бактерии GAS (штамм NZ131) переносили в нейтральный или кислый TSBY для 1-часовой инкубации. МРНК GAS собирали и детектировали с помощью ПЦР в реальном времени с использованием наборов для быстрой ПЦР KAPA SYBR. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. (B и C) Клетки HMEC-1 (B и C) и A549 (C) инфицировали NZ131 дикого типа и speB , удаленным с предварительной обработкой кислотой или без нее. Гентамицин был добавлен для уничтожения внеклеточных бактерий. Был проведен анализ образования колоний, рассчитана кратность репликации GAS и нормализована к количеству интернализованных бактерий через 1 час после инфицирования.Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов.
Вместо подавления экспрессии гена обработка кислотой сильно увеличивала экспрессию гена speB (рис. 4A), и это согласуется с нашими предыдущими данными о том, что низкий pH стимулирует продукцию белка SpeB (43). SpeB, стрептококковый пирогенный экзотоксин, представляет собой цистеиновую протеазу, которая позволяет ГАЗ переваривать ткани хозяина и иммунные медиаторы (45). Недавний отчет показал, что помимо роли в распространении GAS среди тканей хозяина, SpeB также обеспечивает протеазную активность для расщепления рецепторов аутофагии и повышения внутриклеточной выживаемости GAS в эпителиальных клетках (30).Однако наши результаты показали, что обработка с низким pH индуцировала экспрессию speB , но не усиливала внутриклеточный рост GAS в эндотелиальных клетках. Более того, ростовая активность штамма GAS, удаленного из speB , была аналогична активности GAS дикого типа (фиг. 4B). Однако как штаммы дикого типа, так и штаммы, удаленные из speB , подавлялись в эпителиальных клетках (фиг. 4C). Взятые вместе, эти результаты показывают, что предварительная обработка кислотой подавляет экспрессию различных генов вирулентности, но не speB , и что предварительная обработка кислотой отменяет рост GAS в эндотелиальных клетках.
Подкисление ГАЗ-содержащих везикул в эндотелиальных клетках является дефектным. Как правило, везикулярный pH начинает падать сразу после интернализации бактерий в эндосомы или аутофагосомы, и этому способствует последующее слияние с лизосомами, которые обладают более кислым микроокружением. Чтобы исследовать, является ли функция лизосом недостаточной и приводит ли к росту бактерий в эндотелиальных клетках, но не в эпителиальных, мы сначала использовали LAMP-1 в качестве маркера лизосом для наблюдения за расположением лизосом и аутофагосом.Результаты показали, что LAMP-1 совместно локализуется с LC3, маркером аутофагосом, в обоих типах клеток после инфекции GAS (рис. 5). Однако двойные положительные везикулы LC3 и LAMP-1 были увеличены в эндотелиальных клетках по сравнению с эпителиальными клетками через 6 часов после инфицирования. Хотя GAS-индуцированная аутофагия известна своим большим размером аутофагосом по сравнению с канонической аутофагией, эта большая аутофаголизосома не подавляла рост бактерий внутри эндотелиальных клеток. В конечном итоге физической силы репликации GAS, по-видимому, было достаточно, чтобы разорвать поврежденный пузырек (рис.5А, белые стрелки). Кроме того, рост бактерий в эндотелиальных клетках также наблюдался за пределами LC3-положительных везикул, как показано окрашиванием DAPI (фиг. 5A). Напротив, GAS-содержащие LC3-положительные везикулы постепенно исчезали в эпителиальных клетках, указывая на то, что аутофагический поток завершился и что рост GAS был подавлен (фиг. 5B, белые стрелки). Эти данные показывают, что привлечение LAMP-1 и LC3 не обеспечивает такой же супрессивной активности в эндотелиальных клетках, как в эпителиальных клетках.
FIG 5Лизосомы сливаются с LC3-положительными аутофагосомами. (A и B) Клетки HMEC-1 (A) и A549 (B) инфицировали GAS (штамм NZ131) в течение 30 минут, а затем добавляли гентамицин для уничтожения внеклеточных бактерий. Клетки фиксировали и окрашивали антителами против LC3 и против LAMP-1 в указанные моменты времени, а затем наблюдали с помощью конфокальной микроскопии. Прутки, 10 мкм.
Поскольку слияние лизосом может вызывать среду с низким pH в эндосомах или аутофагосомах и способствовать подавлению роста бактерий, возникает один нерешенный вопрос: почему лизосомная функция эндотелиальных клеток недостаточна для очистки GAS.Поэтому мы измерили pH GAS-содержащего, LC3 и LAMP-1 двойного положительного отсека, используя лизотрекер в качестве индикатора низкого pH (pH <5,5). LC3- и LAMP-1-положительные везикулы были сильно декорированы лизотрекером в эпителиальных клетках, но не в эндотелиальных клетках (фиг. 6A, белые стрелки и области, окруженные пунктирной линией). Количественные результаты показали, что ГАС-содержащие аутофагосомы (положительные по LC3) были сильно локализованы с лизосомами (LAMP-1) и что эффективность рекрутирования не различалась между двумя типами клеток.Однако процент положительности лизотрекера с двойным положительным GAS по LC3 и LAMP-1 был намного ниже в эндотелиальных клетках, чем в эпителиальных клетках (фиг. 6B). Ограниченный рост GAS в кислых аутофагосомах был также подтвержден в нормальных эпителиальных клетках почек крысы (NRK) (см. Рис. S4 в дополнительном материале). Эти данные показывают, что низкий pH важен для эпителиальных клеток для подавления роста GAS посредством аутофагии, в то время как потеря сохранения низкого pH в LC3- и LAMP-1-положительных везикулах происходит в эндотелиальных клетках.
Рисунок S4
Рост GAS ограничен кислыми аутофагосомами в эпителиальных клетках NRK. Клетки NRK инкубировали с лизотрекером (75 нМ) в течение 1 ч, а затем инфицировали GAS (штамм NZ131) при MOI 5 в течение 30 мин. Гентамицин был добавлен для уничтожения внеклеточных бактерий. Через 3 часа после инфицирования клетки фиксировали, окрашивали антителом против LC3 и DAPI, а затем наблюдали с помощью конфокальной микроскопии (A). Указанная процентная доля LC3-положительного GAS или LC3- и дважды положительного по лизотрекеру GAS по отношению к общему внутриклеточному GAS.В частности, 84,5% LC3-положительного GAS были положительными по лизотрекеру. В каждом образце наблюдали более 100 клеток, и показаны средние значения ± стандартное отклонение из трех независимых экспериментов (B). Пруток, 10 мкм. Скачать Рисунок S4, файл TIF, 1,2 МБ.Авторские права © 2015 Lu et al.Рис. 6Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Слитые с лизосомами везикулы в эндотелиальных клетках не поддерживают кислую среду. После предварительного окрашивания лизотрекером (75 нМ) в течение 1 часа клетки инфицировали GAS (штамм NZ131) в течение 30 минут, а затем добавляли гентамицин для уничтожения внеклеточных бактерий. Клетки фиксировали и окрашивали антителами против LC3 и против LAMP-1 через 1 и 3 часа после инфицирования. Образцы, собранные через 3 часа после заражения, были визуализированы с помощью конфокальной микроскопии (A). Лизотрекер (LysoT.) Сильно окрашивает LC3- и LAMP-1-положительные везикулы в клетках A549, но не в клетках HMEC-1 (белые стрелки и области, окруженные пунктирной линией).Наблюдалось более 100 клеток в каждом образце, и показаны средние значения ± стандартное отклонение из трех независимых экспериментов (B). Прутки, 10 мкм.
Низкий pH необходим для подавления внутриклеточного роста GAS. Чтобы дополнительно подтвердить, является ли кислый pH ключевым фактором, влияющим на внутриклеточный рост GAS, мы использовали Baf A для блокирования закисления везикул (см. Рис. S5A в дополнительном материале) и подтвердили его нетоксический эффект на рост ГАЗ (рис. S5B). Мы обнаружили, что GAS, подавленный аутофагией, может расти в эпителиальных клетках, обработанных Baf A, что указывает на то, что даже аутофагосомы требуют низкого pH для уничтожения бактерий (рис.7А). Более того, распределение GAS в эпителиальных клетках ограничивалось паттерном L-типа с очень небольшим количеством H-типов, тогда как GAS-инфицированные эндотелиальные клетки демонстрировали распределение H-типа (фиг. 7B). Взятые вместе, эти данные показывают, что поддержание среды с низким pH в везикулах важно для внутриклеточного подавления роста GAS.
Рисунок S5
Бафиломицин A1 подавляет внутриклеточное закисление, но не влияет на рост GAS. (A) Клетки A549 инкубировали с Baf A (100 нМ) и лизотрекером (75 нМ) в течение 1 часа.Клетки фиксировали и окрашивали антителом против LAMP-1 и DAPI, а затем наблюдали под конфокальной микроскопией. Пруток, 10 мкм. (B) После ночного культивирования бактерии GAS (штамм NZ131) переносили в свежий бульон TSBY, содержащий Baf A (100 нМ) или гентамицин (100 мкг / мл). Рост бактерий измеряли при OD 600 в различные моменты времени. Данные представляют собой средние значения ± стандартное отклонение от трех культур. Опыты повторяли дважды. Скачать Рисунок S5, файл TIF, 0,9 МБ.Авторские права © 2015 Lu et al.Рис. 7Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Низкое значение pH в эпителиальных клетках необходимо для подавления репликации GAS. (A) Клетки HMEC-1 и A549 с или без 1-часовой предварительной обработки бафиломицином A1 (100 нМ) инфицировали GAS (штамм NZ131) в течение 30 минут, а затем обрабатывали гентамицином для уничтожения внеклеточных бактерий.Бактерии собирали в указанные моменты времени после обработки гентамицином, рассчитывали кратность репликации GAS и нормировали на количество интернализованного GAS через 0,5 часа после заражения. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. (B) ГАЗ H-типа, как описано в легенде к фиг. 3, измеряли в инфицированных клетках HMEC-1 и A549. Процент клеток с инфекцией GAS и клеток с рисунком H-типа рассчитывали с помощью конфокальной микроскопии. Наблюдалось более 100 клеток в каждом образце, и показаны средние значения ± стандартное отклонение из трех независимых экспериментов.**, P <0,01.
ОБСУЖДЕНИЕ
В настоящем исследовании низкий pH идентифицирован как главный фактор в снижении репликации GAS во внутриклеточной среде. Процесс везикулярного подкисления строго регулируется в клетках, и его компартментализация в везикулах способствует внутриклеточному бактериальному клиренсу. В эпителиальных клетках аутофагосомы поглощают ГАЗ, чтобы обеспечить неповрежденный компартмент для поддержания низкого pH и функции лизосом. Рост бактерий может быть подавлен посредством эндосомной деградации, если GAS предварительно обработали средой с низким pH перед интернализацией.В эндотелиальных клетках, однако, подкисление не поддерживается внутри GAS-содержащих везикул, хотя маркеры LC3 и LAMP-1 легко рекрутируются в них. Наши результаты подтверждают необходимость низкого pH для подавления внутриклеточного роста GAS в эпителиальных клетках посредством аутофагии, в то время как дефектное закисление позволяет репликации GAS в эндотелиальных клетках (рис. 8).
FIG 8Влияние pH на рост GAS в эндотелиальных и эпителиальных клетках. В эндотелиальных клетках GAS интернализуется в клетки посредством эндоцитоза и покидает эндосомы, секретируя факторы вирулентности, такие как SLO, которые разрушают структуру мембраны.Аутофагия частично украшает GAS маркерами LC3 и LAMP-1, но это не сохраняет среду с низким pH, и GAS реплицируется внутри эндотелиальных клеток. В эндотелиальных клетках аутофагия неэффективна. Предварительная обработка кислотой снижает вирулентность и рост GAS до инфицирования, а после интернализации подкисление эндосом может подавлять репликацию GAS за счет эндосомной деградации. С другой стороны, в эпителиальных клетках аутофагия обеспечивает лучшее сохранение низкого pH и эффективно ограничивает и убивает бактерии.
После того, как GAS-содержащие эндосомы созревают в поздние эндосомы или GAS окружается аутофагосомой, pH внутри начинает снижаться в любом случае и способствует слиянию с лизосомами (26, 46). Хотя наши результаты показывают, что LAMP-1 пометил LC3-позитивные везикулы, мы не можем быть уверены, были ли лизосомы, в частности, уже слились с ними или нет, потому что LAMP-1 является маркером как для лизосом, так и для поздних эндосом. Однако независимо от того, на какой стадии созревания находятся эти везикулы, (т.е., на поздних эндосомах, аутофаголизосомах или лизосомах) для нормальной обработки требуется низкий pH. Наиболее поразительно то, что в эндотелиальных клетках не поддерживался компартмент с низким pH, и это приводило к отсутствию подавления роста GAS.
Потеря низкого pH могла быть вызвана дефектом транспорта протонов из цитоплазмы в пузырьки или утечкой протонов из пор, образованных факторами вирулентности бактерий. Хотя мы обрабатывали клетки Baf A, который специфически блокирует перенос протонов из цитоплазмы в везикулы и отменяет ингибирующую активность репликации GAS в эпителиальных клетках, точный механизм потери протонов из везикул в GAS-инфицированных клетках остается неясным.Предыдущие сообщения показали, что потеря подкисления отменяет созревание фагосом и что обработка Baf A может повлиять на слияние лизосом с аутофагосомами (47). Однако мы обнаружили, что LAMP-1 локализован на аутофагосомах с нейтральным pH в эндотелиальных клетках, что указывает на то, что эти события могут быть несвязанными.
Сообщалось, что изменение pH влияет на транскрипцию генов факторов вирулентности, включая slo , sagB и mga GAS в стационарной фазе, а также может иметь отношение к фазе роста бактерий (40).Экспрессия гена mga регулирует поверхностные белки GAS, контролирует колонизацию GAS и нарушает закисление фагосом в клетках сосудистой системы (48, 49). Мы также продемонстрировали, что простая обработка средой с низким pH может снизить экспрессию гена фактора вирулентности GAS, и это важно для внутриклеточного выживания GAS. Это означает, что раннее закисление может быть защитным механизмом хозяина для снижения экспрессии генов slo , sagB , spn ( nga ) и mga и уменьшения их вирулентного воздействия на разрыв мембраны и закисление фагосом.Это предполагает, что может существовать конкуренция между активацией фактора вирулентности GAS и закислением везикул хозяина. Недавний отчет показал, что SLO и NAD-гликогидролаза предотвращают закисление фаголизосом и способствуют выживанию GAS в макрофагах (35). Следовательно, существует сложное взаимодействие между факторами вирулентности бактерий и компонентами аутофагии (50). Следовательно, эпителиальные клетки быстро реагируют на эндосомные повреждения, задействуя аутофагический аппарат для прямого захвата GAS или даже GAS-содержащей эндосомы внутри аутофагосомы до образования достаточно большого разрыва в эндосомальной мембране, который позволит GAS или протонам ускользнуть.Этот процесс очень эффективен для подавления роста ГАЗА на самых ранних стадиях инфекции, а также подчеркивает важность поддержания кислотного компартмента как жизненно важного первого шага в борьбе с вторжением бактерий, которые могут повредить мембраны.
Однако предыдущее сообщение показало, что SpeB расщепляет рецепторы аутофагии и способствует уходу M1T1 GAS от аутофагии в эпителиальных клетках (30). В этом исследовании рост внутриклеточного GAS был только примерно в два раза выше у дикого типа, чем у M1T1 GAS, удаленного из speB .Возможные причины наблюдаемых различий в двух исследованиях могут включать различия во временных точках бактериального анализа (2, 4 и 6 ч против 8 ч после заражения). Наши результаты не исключают возможную роль SpeB во внутриклеточной защите от аутофагического уничтожения эпителиальных клеток, но указывают на то, что такая роль может быть относительно незначительной.
Один из использованных нами штаммов GAS, A20, также является штаммом серотипа M1T1, который был выделен из крови пациента с сепсисом и имеет фенотип с высокой экспрессией SpeB.Однако мы обнаружили, что штамм A20 не может выжить и расти в эпителиальных клетках (см. Рис. S1 в дополнительном материале). Напротив, A20 мог реплицироваться в эндотелиальных клетках, и количество бактерий было в 10 раз выше через 6 часов после инфицирования, чем количество интернализованных бактерий (рис. 1A). Мы также показали, что не было никакой разницы в количестве бактерий между NZ131 дикого типа и SW574, удаленным по speB , в эндотелиальных клетках (фиг. 4C). Кроме того, предварительная обработка кислотой, которая увеличила экспрессию speB (рис.4A), не способствовал внутриклеточной выживаемости GAS (фиг. 4B). Напротив, SLO требуется для внутриклеточного выживания GAS, тогда как GAS, предварительно обработанный кислотой, и GAS, удаленный slo, , снижали способность GAS к росту в эндотелиальных клетках. Наши результаты показывают, что рост GAS является фенотипом, зависимым от типа клеток, и что во внутриклеточном росте бактерий участвуют различные факторы вирулентности, но не SpeB.
Предварительная обработка кислотой и среда в стационарной фазе могут рассматриваться как аналогичные ситуации, обнаруженной в эпидермисе, где колонизирующие ГАЗ сталкиваются с ограниченными питательными веществами и постепенно снижающимся pH окружающей среды и накоплением продуктов метаболизма.Воспользовавшись снижением экспрессии генов вирулентности при измерении pH с помощью GAS, кератиноциты и эпителиальные клетки могут легко контролировать и очищать интернализованный GAS, особенно по сравнению с эндотелиальными клетками, которые сталкиваются с GAS в условиях нейтрального pH (28, 34). Как только ГАЗ распространяется в систему кровообращения при сепсисе, нейтральный pH и энергетическое богатство сыворотки побуждают ГАЗ вернуться к более опасной фазе экспрессии генов, которая может вызвать большее повреждение кровеносных сосудов. Используя модель инфекции воздушного мешка, установленную ранее (51), наши предварительные результаты показывают, что мыши более устойчивы к ГАЗ в стационарной фазе, чем к ГАЗ в ранней логарифмической фазе (данные не показаны).
Эпителиальные и эндотелиальные клетки обладают разными физиологическими функциями. Эпителиальные клетки обычно выстилают внешние поверхности, которые предназначены для взаимодействия с инородными материалами и обеспечения защиты от патогенов. Напротив, эндотелиальные клетки обычно занимают внутреннюю, стерильную и богатую питательными веществами среду, что может потребовать менее строгих аутофагических реакций. Сильный аутофагический ответ (например, в эпителиальных клетках) способен подавлять внутриклеточный рост GAS, тогда как дефектный аутофагический ответ, такой как наблюдаемый в эпителиальных клетках с нокаутом atg7 , приводит к плохому уничтожению GAS и позволяет репликацию GAS (см.рис.S6A и B в дополнительном материале). Это указывает на то, что помимо различий в физиологических функциях эпителиальных и эндотелиальных клеток, аутофагия является ключевым шагом к поддержанию низкого pH в лизосомном / аутофагосомном компартментах и подавлению внутриклеточного роста GAS. Кроме того, мы подтвердили, что, стандартизованные по размеру клеток, исходные средние уровни pH клеток подобны для этих двух типов клеток (рис. S7). Однако неэффективная аутофагия в эндотелиальных клетках вызывает потерю способности сохранять низкий pH в GAS-содержащих аутофагосомах, что приводит к репликации GAS.Наиболее важно то, что мы показали, что как линия эндотелиальных клеток (HMEC-1), так и первичные эндотелиальные клетки (эндотелиальные клетки пупочной вены человека [HUVECs]; Рис. S8) неспособны очищать внутриклеточный ГАЗ из-за недостаточного подкисления во время процесса аутофагии. В будущем будет интересно изучить другие типы эндотелиальных клеток на предмет их реакции на инфекцию GAS.
Фигура S6
Рост GAS подавляется в эпителиальных клетках дикого типа, но не в эпителиальных клетках atg7 , нокаутированных по A549.(A) Клетки дикого типа A549 и atg7 , полученные с помощью кластеризованной системы коротких палиндромных повторов с регулярными интервалами между ними (CRISPR) -Cas9, были получены от Tamotsu Yoshimori. Клетки обрабатывали средой EBSS с или без Baf A (100 нМ) в течение 2 ч и собирали для определения белка Atg7 (013-22381; Wako) и LC3 (PM036; MBL) вестерн-блоттингом. Поток аутофагии блокировался в atg7 -нокаутных клетках A549. (B) Клетки инфицировали GAS (штамм NZ131) при MOI 1 или 5 для клеток HMEC-1 и A549, соответственно, в течение 30 минут, и внеклеточные бактерии были убиты обработкой гентамицином.Количество внутриклеточных бактерий определяли с помощью колониеобразующего анализа через 1 и 6 часов после заражения. Подсчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий через 1 час после инфицирования. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. Скачать Рисунок S6, файл TIF, 0,3 МБ.Авторские права © 2015 Lu et al.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Рисунок S7
Сравнение уровней pH между клетками HMEC-1 и A549. Клетки высевали для ночного культивирования. Клетки без фиксации окрашивали акридиновым оранжевым (A.O.) (5 мкг / мл) в течение 10 мин при 37 ° C. Среднюю интенсивность флуоресценции размеров клеток (прямой рассеянный свет [FSC]; площадь или размер клеточной поверхности) (A) или окрашивание АО (B) анализировали с помощью проточной цитометрии. Данные представляют собой средние значения ± стандартное отклонение от трех независимых экспериментов. Скачать Рисунок S7, файл TIF, 0,1 МБ.Авторские права © 2015 Lu et al.Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Unported, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Рисунок S8
GAS реплицируется в эндотелиальных клетках пупочной вены человека (HUVEC). Клетки HUVEC инфицировали GAS (штамм NZ131) при MOI 1 в течение 1 часа. Внеклеточные бактерии были убиты обработкой гентамицином.Количество интернализованных бактерий определяли с помощью анализа образования колоний в различные моменты времени после заражения. Подсчитывали кратность репликации GAS и нормировали на количество интернализованных бактерий через 2 часа после инфицирования. Данные представляют собой средние значения ± стандартное отклонение от трех культур. Опыты повторяли дважды. Скачать Рисунок S8, файл TIF, 0,1 МБ.Авторские права © 2015 Lu et al.Это статья в открытом доступе, распространяемая на условиях Creative Commons Attribution-Noncommercial-ShareAlike 3.0 Непортированная лицензия, которая разрешает неограниченное некоммерческое использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Низкий pH не только напрямую влияет на вирулентность бактерий, он также влияет на реакцию хозяина, поскольку низкий pH жизненно важен для активности деградирующих ферментов в лизосомах (25). Механизм того, как клетка поддерживает кислую среду в определенном месте, таком как поздние эндосомы, аутофагосомы и лизосомы, является важным вопросом клеточной биологии.Здесь мы показываем, что низкий уровень pH поддерживается за счет использования аутофагического механизма, который позволяет клеткам защищаться от внутриклеточного ГАЗ. Подробные молекулярные механизмы относительно того, как GAS может неконтролируемым образом реплицироваться в эндотелиальных клетках, требует дальнейшего изучения. Недавние сообщения показали, что ассоциация фагосом с LC3, в отсутствие изоляционной мембраны, необходима для созревания фагосом и последующего снижения pH и рекрутирования кислых гидролаз. Связанные с LC3 фагосомы сливаются с лизосомами и инактивируют вторгшиеся патогены (52).Не сообщалось, требуется ли ассоциация LC3 для успешного уничтожения бактерий в нефагоцитах. Выключение основных аутофагических белков, включая FIP200 и Atg9, в клетках эмбриональных фибробластов мыши привело к отсутствию образования изоляционной мембраны и подавлению роста бактерий, даже несмотря на то, что LC3 был задействован в эндосомах, содержащих Salmonella (53). Мы предполагаем, что отсутствие изоляционной мембраны в эндотелиальных клетках, даже если LC3 все еще был обнаружен на GAS-содержащих везикулах, может привести к репликации GAS внутри разорванной мембраны и последующему росту в цитоплазме.Эту гипотезу еще предстоит подтвердить.
В заключение, низкий pH напрямую влияет на рост GAS в культуральной среде и необходим для подавления аутофагии репликации GAS внутри клеток. Результаты, полученные в результате этого исследования, могут помочь лучше понять и предоставить потенциальные терапевтические стратегии при заболеваниях ГАЗ.
МАТЕРИАЛЫ И МЕТОДЫ
Культура клеток. Линия 1 эндотелиальных клеток микрососудов человека (клетки HMEC-1), полученная из Центров по контролю и профилактике заболеваний, США, выращивалась в культуральных планшетах, содержащих среду для роста эндотелиальных клеток M200 (Cascade Biologics) состоит из 10% фетальной бычьей сыворотки (FBS), 1 мкг / мл гидрокортизона, 10 нг / мл эпидермального фактора роста, 3 нг / мл основного фактора роста фибробластов и 10 мкг / мл гепарина.Клетки HMEC-1 сохраняют морфологические, фенотипические и функциональные характеристики нормальных эндотелиальных клеток микрососудов человека (54). Эпителиальные клетки карциномы легкого человека A549 и нормальные эпителиальные клетки почек крысы (NRK) поддерживали в среде Игла, модифицированной Дульбекко (DMEM), с добавлением 10% FBS. Клетки культивировали при 37 ° C в 5% CO 2 и отделяли 1000 ед / мл трипсина и 0,5 мМ EDTA для пассажа. Когда конфлюэнтность клеток достигала 80%, клетки отделяли трипсином-ЭДТА и высевали 1 × 10 6 клеток в 10-сантиметровые чашки для поддержания, 2 × 10 5 клеток в 6-луночные планшеты для анализа проточной цитометрии. 8 × 10 4 клеток в 24-луночных планшетах для анализа образования колоний и 6 × 10 4 клеток в 24-луночных планшетах с покровным стеклом для наблюдения под флуоресцентным микроскопом.Для ингибирования подкисления клетки предварительно обрабатывали бафиломицином A1 (номер по каталогу B1793; Sigma) в концентрации 100 нМ в течение 1 ч, а затем инфицировали стрептококком группы A (GAS), как описано в протоколе заражения.
Bacteria.GAS штамм NZ131 (серотип M49) был подарком от Д. Р. Мартина (Центр инфекционных болезней Новой Зеландии, Порируа), а штамм SW574, удаленный speB, был описан ранее (55). A20 (серотип M1T1), SW555 (штамм A20, удаленный slo ), JRS4 (серотип M6) и штаммы JRS4, удаленный slo , были от J.J. Wu (Департамент медицинских лабораторных исследований и биотехнологии, Медицинский колледж Национального университета Ченг Кунг, Тайнань, Тайвань) и другие клинически изолированные штаммы были получены от CC Liu (Департамент педиатрии, Медицинский колледж Национального университета Ченг Кунг, Тайнань, Тайвань) . Все эти штаммы GAS были изолированы от пациентов с GAS-инфицированными инвазивными заболеваниями, такими как сепсис, синдром токсического шока стрептококка (STSS), гломерулонефрит и некротический фасциит. GAS вырос при 37 ° C в триптическом соевом бульоне, содержащем 0.5% дрожжевой экстракт (TSBY) в течение ночи и переносили в свежий бульон при разведении 1:50 на 3 часа (свежий ГАЗ). Обновленный GAS, ранний журнал GAS, использовался в качестве стандартной культуры для экспериментов. Бактерии собирали центрифугированием (3500 об / мин, 10 мин, 4 ° C), ресуспендировали в фосфатно-солевом буфере (PBS) и определяли концентрацию, используя оптическую плотность при 600 нм (OD 600 ) 0,2 как 1. × 10 8 КОЕ / мл и подтверждено подсчетом жизнеспособных колоний.
Определение pH.После роста GAS в течение различного времени супернатант бактериальной культуры собирали центрифугированием и затем пропускали через фильтр 0,22 мкм. Значения pH измеряли с помощью pH-метра (F-52; Horiba). Для экспериментов по ингибированию кислоты pH свежего бульона доводили соляной кислотой (HCl) и добавляли GAS к культуре в течение указанных периодов времени.
Модель инфекции. Клетки с 80% конфлюэнтностью помещали в 24-луночные планшеты или 6-луночные планшеты и инкубировали в течение ночи. Подготовленные бактерии с предварительной обработкой кислой среды в течение 1 ч или без нее добавляли непосредственно в лунки при различной множественности инфекции (MOI).Для одновременного инфицирования клеток планшеты центрифугировали при 500 × g в течение 5 мин при 4 ° C. После 30-минутной или 1-часовой инкубации клеточную культуру трижды промывали PBS и добавляли свежую среду, содержащую 100 мкг / мл гентамицина, для уничтожения внеклеточных бактерий. Клетки собирали в различные периоды времени, как указано в каждом эксперименте.
Анализ проточной цитометрии для внутриклеточного окрашивания GAS. Клетки высевали при 2 × 10 5 в 6-луночные планшеты для ночного культивирования и инфицировали GAS при MOI 25 в течение 1 часа.Гентамицин был добавлен для уничтожения внеклеточных бактерий. Клетки собирали трипсинизацией и фиксировали 4% параформальдегидом, повышали проницаемость 0,1% сапонином в PBS, содержащем 2% FBS, и окрашивали кроличьей поликлональной анти-GAS антисывороткой в разведении 1: 10000 в течение 1 ч, которые были созданы в лаборатории J. J. Wu. После окрашивания вторичных антител, конъюгированных с Alexa Fluor 488, образцы анализировали с помощью проточной цитометрии (FACSCalibur; BD Biosciences). Окрашивание только вторичным антителом использовали в качестве фонового контроля.
ПЦР в реальном времени.GAS РНК выделяли, как описано ранее (56). После ПЦР с обратной транскриптазой кДНК измеряли с помощью ПЦР в реальном времени в соответствии с наборами для быстрой ПЦР KAPA SYBR (KAPA Biosystems, Woburn, MA). Олигонуклеотидные последовательности праймеров были следующими: 5′-TCCTGCGGATGTGTTTGATA-3 ‘и 5′-TGCACTAAAGGCCGCTTC-3’ для slo , 5′-CTGTGCTTCAGGTGGAGGTTTAATT-3 ‘и 5’-ATAGCACGCG для ′ -TGTTGCTATTGCTTTGGCTG-3 ′ и 5′-TTGAGCCGTCTAATGTGTGC-3 ′ для spn ( nga ), а также 5′-AATTGATGGCTGATGTTGGTAT-3 ′ и 5T′-GCTTCCCAATGCTGCTG spear. [ΔCP цель (контроль — образец) — ΔCP ссылка (контроль — образец)] (57).
Иммунофлуоресцентное окрашивание. Клетки высевали при 6 × 10 4 в 24-луночные планшеты с покровным стеклом для ночного культивирования и инфицировали GAS в течение 30 мин. Для окрашивания кислотным индикатором добавляли лизотрекер (красный DND-99; Invitrogen) в конечной концентрации 75 нМ в течение 1-часовой инкубации перед заражением GAS. Внеклеточные бактерии были убиты гентамицином в дозе 100 мкг / мл. В различные моменты времени после инфицирования клетки фиксировали 4% параформальдегидом, пермеабилизировали 50 мкг / мл дигитонина и окрашивали антителами против LC3 (pM036; MBL) и против LAMP-1 (h5A3; Санта-Крус) при комнатной температуре. за 1 ч.После промывки клеток PBS их окрашивали вторичными антителами, конъюгированными с Alexa Fluor, и DAPI в течение 40 мин, а затем образцы анализировали с помощью конфокальной микроскопии (FV1000; Olympus).
Статистический анализ. Статистический анализ проводился с использованием парного теста t . Значимые различия были установлены при значениях P <0,05 (версия программного обеспечения GraphPad Prism 5).